Abstract
Prions are self-templating protein aggregates that were originally identified as the causative agent of prion diseases in mammals, but have since been discovered in other kingdoms. Mammalian prions represent a unique class of infectious agents that are composed of misfolded prion protein. Prion proteins usually exist as soluble proteins but can refold and assemble into highly ordered, self-propagating prion polymers. The prion concept is also applicable to a growing number of non-Mendelian elements of inheritance in lower eukaryotes. While prions identified in mammals are clearly pathogens, prions in lower eukaryotes can be either detrimental or beneficial to the host. Prion phenotypes in fungi are transmitted vertically from mother to daughter cells during cell division and horizontally during mating or abortive mating, but extracellular phases have not been reported. Recent findings now demonstrate that in a mammalian cell environment, protein aggregates derived from yeast prion domains exhibit a prion life cycle similar to mammalian prions propagated ex vivo. This life cycle includes a soluble state of the protein, an induction phase by exogenous prion fibrils, stable replication of prion entities, vertical transmission to progeny and natural horizontal transmission to neighboring cells. Our data reveal that mammalian cells contain all co-factors required for cytosolic prion propagation and dissemination. This has important implications for understanding prion-like properties of disease-related protein aggregates. In light of the growing number of identified functional amyloids, cell-to-cell propagation of cytosolic protein conformers might not only be relevant for the spreading of disease-associated proteins, but might also be of more general relevance under non-disease conditions.
Keywords: :
Prions—Self-Templating Protein Conformers with Infectious Properties
Prions were originally identified as the causative agent of the transmissible spongiform encephalopathies (TSE) in humans and other mammals.Citation1 Prion diseases include scrapie in sheep and goats, chronic wasting disease in deer, elk and moose, bovine spongiform encephalopathies and human prion diseases such as Creutzfeldt-Jakob disease, Gerstmann-Sträussler-Scheinker disease and fatal familial insomnia. Prion diseases typically occur sporadically or are of genetic origin. Importantly, prion diseases can also be acquired and can sometimes even be transmitted across species barriers. The term prion was first introduced by Stanley Prusiner to describe a novel class of “proteinaceous infectious particles” devoid of coding nucleic acids.Citation1 According to the protein-only hypothesis, mammalian prions consist of cellular prion protein PrP that gains infectious properties by a conformational transition into an aggregated, β-sheet rich prion isoform, termed PrPSc. The precursor of mammalian prions is the cellular prion protein, PrPC. PrPC is a highly glycosylated, gycosylphosphatidyl-inositol anchored cell-surface protein abundantly expressed in the central nervous system but also in other tissues.Citation2,Citation3 Mammalian prion formation is confined to the cell surface or cytosolic vesicles upon direct contact of the prion conformer PrPSc with PrPC (for review, see ref. Citation4).
The self-perpetuating changes in protein conformation are also a hallmark of several proteins found in lower eukaryotes such as yeast and filamentous fungi. Fungal prion proteins share little sequence similarity with mammalian PrP. One exception is the translation termination factor Sup35 of Saccharomyces cerevisiae (S. cerevisiae), that harbors a peptide repeat region similar to the one in PrP.Citation5,Citation6 Prion proteins of lower eukaryotes can also form insoluble, self-perpetuating amyloid-like polymers in vitro and in vivo.Citation7 Yeast prions constitute protein-based epigenetic elements that confer heritable phenotypic traits to their hosts.Citation8,Citation9 At least nine different prion proteins of fungi have been identified that can be lethal or beneficial to the host.Citation10-Citation12 Yeast prions are predominately cytoplasmic, with one exception that resides within the nucleus.Citation13 Thanks to the power of yeast genetics and the establishment of tractable systems to monitor prion states, yeast has proven to be a versatile model system to study mechanistic details of protein-based inheritance. Importantly, however, it must not be forgotten that mammalian and yeast prions differ in their cellular localization. Consequently, the exact factors required for prion induction and propagation for fungal and mammalian prions likely differ.
Mammalian and yeast prions replicate by a seeded-polymerization mechanism in which misfolded prion protein catalyzes the conformational switch of the soluble protein and its incorporation into highly-ordered fibrillar aggregates, so-called amyloid. For multiplication of misfolded protein conformers, secondary nucleation events are required that generate daughter seeds.Citation14-Citation17 Secondary nucleation is an essential step in the life cycle of prions. Results obtained with yeast prions highlight the essential role of chaperone-mediated aggregate shearing for prion maintenance.Citation18-Citation20 During yeast budding, prion seeds transmit to the daughter cells where they recruit soluble prion proteins and grow into longer filaments. Severing of prion fibrils by cellular chaperones then generates fibril fragments that restore the mean prion population before cytokinesis is initiated again. In mammals, prion seed multiplication is required for efficient dissemination within and between organisms, but co-factors involved in this process have not been revealed so far. Under rare circumstances, prion seeds can form spontaneously, giving rise to sporadic prion diseases in humans or heritable phenotypic traits in yeast. The mechanism of spontaneous prion induction in mammals is not understood, but evidence from yeast prions demonstrates that other ordered protein polymers or even prions can theoretically act as heterologous seeds that cross-seed protein misfolding and thus induce heritable prion states.Citation21-Citation23 As spontaneous formation of PrP-derived prions has so far not been observed in cellular models (for review see ref. Citation4), prions of lower eukaryotes serve as important models for studying cellular aspects of spontaneous prion induction.
Prions induce self-perpetuating prion states when transmitted to other cells. Cytoplasmic yeast prions are transmitted vertically to progeny or horizontally during mating.Citation24 Yeast prions can also be transmitted experimentally by cytoduction, a process of abortive mating, resulting in cytoplasmic mixing without nuclei fusion.Citation8 Extracellular phases in the yeast prion replication cycle have not been observed.Citation25 Prions in mammals follow a complex life cycle that enables horizontal transmission between individuals of the same species and even across species barriers. From the initial site of infection, prions spread within the host to eventually target the central nervous system, where they multiply to high levels.Citation26 The exact mechanism of intercellular spreading of mammalian prions is not fully understood, but cell culture models have shed some light onto the cellular mechanisms that might be involved also in vivo. Mammalian prions ex vivo transmit horizontally to bystander cells, in which they induce sustained infection (for a review, see ref. Citation4). In some models, direct contact between cells ex vivo is required.Citation27 Cytoplasmic bridges such as tunneling nanotubes (TNTs) appear to be involved in mammalian prion spreading ex vivo.Citation28 TNTs represent special types of intercellular bridges that allow intercellular exchange of cytoplasm and even organelles such as lysosomes and mitochondria.Citation29 Dissemination of mammalian prions ex vivo also includes extracellular phases. Mammalian prions have been found associated with exosomes, and those exosome fractions effectively induce prion infection in tissue culture and animal models.Citation30-Citation33 It has been hypothesized that the efficient dissemination of mammalian prions is directly related to the membrane association of PrP, facilitating interaction with necessary co-factors for propagation and cell-to-cell spreading by membrane-bound vesicles or intercellular membrane interactions.Citation34 So is prion biogenesis in mammalian cells unique to PrP and its localization on the cell surface?
Prion Propensity of a Yeast Prion Domain in the Mammalian Cytosol
Recent findings now argue that the cytosol of mammalian cells also supports aggregate propagation and that membrane anchorage is not a prerequisite for prion replication.Citation35,Citation36 Evidence for this comes from experiments with mammalian cell cultures ectopically expressing the prion domain of the S. cerevisiae Sup35 protein, a subunit of the translation termination factor. In yeast, prions composed of aggregated Sup35 cause a nonsense suppression phenotype due to occasional read-through of stop codons.Citation9 Sup35 is a 686 amino acid residue protein comprising domains N, M, and C domain. Prion propensity of Sup35 is governed by a transposable N-terminal (N) and middle domain (M). The N domain is enriched in glutamine and asparagine residues and harbors a peptide repeat domain. N is required to form the self-propagating amyloid in yeast.Citation37-Citation39 The highly charged middle domain (M, aa 125–253) enhances the protein solubility and is required for sustained mitotic and meiotic stability of the prion phenotype.Citation40 The C-terminal domain (C, aa 254–686) has translation termination activity in yeast and is dispensable for prion formation.Citation40
To elucidate if the mammalian cytosol can also support prion-like propagation of protein aggregates, the Sup35 NM prion domain was stably expressed in murine cells and its prion propensity was assessed upon induction with exogenous NM aggregate seeds. As the NM domain by itself does not normally exist in mammalian cells, it allows monitoring cytosolic prion propagation without loss-of-function phenotype. Cytosolic prion propagation was initially studied in the mitotically active mouse neuroblastoma cell line N2a and subsequently also in primary neurons and astrocytes () and organotypic hippocampal slices.Citation35,Citation41 In all models, ectopically expressed cytosolic NM remained soluble and did not significantly affect cell viability. Remarkably, experimentally induced raise in reactive oxygen species which trigger aggregation of other amyloidogenic proteins, did not affect solubility of the NM domain in neuroblastoma cells.Citation41 Even upon continuous culture, spontaneous induction of NM prion aggregates was not observed, demonstrating that Sup35 NM is not prone to spontaneously misfold when cytosolically expressed. To induce aggregation of Sup35 NM in mammalian cells, we employed previously established prion induction protocols using recombinant yeast prion protein fibrils.Citation42-Citation45 In yeast, the prion phenotype can be induced by transformation of recombinant Sup35 NM fibrils via liposomes or polyethylene glycol (PEG).Citation42,Citation43,Citation45 Also in mammalian cells, exogenously added recombinant NM fibrils were efficiently taken up by cells and induced aggregation of the ectopically expressed soluble NM.Citation35 The mechanism of NM internalization is unclear, but pathways involved in the uptake of PrP-derived prions or other amyloidogenic protein aggregates, such as endocytosis or macropinocytosis might play a role,Citation46-Citation51 potentially after binding to specialized membrane regions enriched in cholesterol.Citation52
Figure 1. Confocal image of an astrocyte transduced with NM-GFP encoding lentivirus. The cells were treated with 1 µM recombinant NM fibrils (monomer equivalent) for 24 h and fixed 48 h post fibril addition. A net-like NM-GFP aggregate structure with very high fluorescence intensity was visible close to the nucleus (left). Higher magnification of the cell periphery showed further spindle-shaped NM-GFP (green) aggregates present throughout the cytoplasm (right). Nuclei were stained with Hoechst (blue) and GFAP with anti-GFAP antibody (red). (Scale bar = 5 µm).
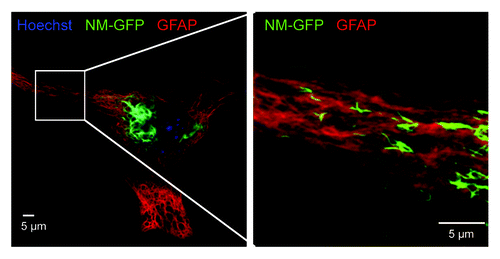
Once induced, Sup35 NM prion aggregates were remarkably stable.Citation35 Even under prolonged cell culture over months, the prion phenotype was faithfully maintained. Self-templating aggregates were also induced in a PrP-deficient cell line stably expressing NM, demonstrating that the cellular prion protein is not involved in NM prion induction and propagation.Citation36 NM aggregates differed in morphology, ranging from small punctate aggregates to longer, worm-like aggregates that were maintained even upon cell division. Subsequent isolation of N2a clones by limiting dilution cloning confirmed that aggregate phenotypes were inherited by daughter cells, arguing that aggregates had self-propagating properties. Surprisingly, N2a cells appeared to be unaffected by the continuous presence of NM aggregates, as significant changes in cell viability were not observed.Citation41 Again, continuous culture of several clones did not lead to a loss of the prion phenotype, and even years after the first induction, more than 90% of the cells in closed populations contain NM prions. This is in line with mammalian prion propagation in mitotically active tissue culture cells.Citation4 Ex vivo propagation of mammalian prions is generally not cytotoxic, and sustained propagation ex vivo over years has been achieved for several mouse-adapted prion strains.Citation53-Citation55
But how exactly can protein aggregates propagate in mammalian cells? Clearly, the stable propagation of mammalian prions in dividing cells argues that aggregate propagation is quite efficient, because the prion multiplication effectively outcompetes prion loss due to cell division or clearance. Secondary nucleation events that produce more seeds from a preformed seed and bidirectional seed distribution to both daughter cells are key characteristics of yeast and mammalian prions in mitotically active cells.Citation24,Citation36,Citation56 Cells are equipped with sophisticated proteostasis mechanisms that sense, refold or ultimately degrade aberrantly folded proteins. As misfolded proteins can be cytotoxic and thus pose a potential risk to cells, damaged and misfolded proteins are often retained by the mother cell during cytokinesis to increase fitness of progeny.Citation57,Citation58 Proteins that can function as prions appear to have evolved mechanisms that allow them to sustain complete clearance by cellular degradation machineries. Still, interaction of prions with the cellular protein folding and clearance machinery can also aid in prion propagation. Yeast prions crucially depend on the cellular chaperone system for seed multiplication. A major player in yeast prion propagation is the AAA+ ATPase Hsp104 capable of resolubilizing proteins and rapidly dissociating protein aggregates.Citation59,Citation60 Hsp104 shears high molecular weight prion aggregates into smaller seeds,Citation18 a process that is tightly regulated by the interplay of Hsp104 with chaperones Hsp70 and Hsp40.Citation20,Citation61,Citation62 Experimental evidence suggests that the rate of aggregate fragmentation is dictated by fibril properties and host cell environment and is a key determinant for transmissibility.Citation16 The effective shearing of growing prion aggregates thus enables sustained prion propagation in rapidly dividing populations.Citation18,Citation63-Citation65 Importantly, aggregate shearing results in multiplication of mammalian prions in a test tube in vitro, suggesting that aggregate fragmentation by host factors might also contribute to mammalian prion replication in vivo.Citation66,Citation67 While processes that support secondary seed formation in mammalian prion replication are unknown, the propagation mechanism of cytosolic NM prions likely differs from that of mammalian prions that replicate along the endocytic pathway. As the prion disaggregase Hsp104 necessary for prion maintenance in yeast lacks mammalian homologs,Citation68 other cellular factors or processes must trigger cytosolic aggregate shearing in mammalian cells. First evidence for a protein depolymerizing system in mammals came from studies with mammalian cell extracts revealing an ATP-dependent disaggregating activity capable of dissolving preformed protein aggregates and even amyloid fibrils.Citation69,Citation70 Hsp70 and Hsp40 are also key chaperones in mammals that prevent protein misfolding and promote refolding into functional proteins. Recent studies now demonstrate that cooperation with specific cochaperones confers disaggregation activity to the Hsp70-Hsp40 chaperone system.Citation70,Citation71 Hsp110 proteins represent a subgroup of the Hsp70 superfamily that function as nucleotide exchange factors, stimulating ADP release and substrate release by chaperone Hsp70.Citation72,Citation73 Hsp110Citation74,Citation75 synergizes with Hsp70 and Hsp40 to slowly disaggregate protein aggregates.Citation70,Citation71 Chaperones Hsp70, Hsp40, and Hsp110 also stimulate NM fibril disassembly in vitro,Citation76 and could thus also be involved in NM protein polymer fragmentation in mammalian cells. Alternatively, the autophagy machinery could contribute to prion seed formation, as has been suggested for mammalian prions.Citation77,Citation78 Of note, Sup35 NM aggregates can associate with autophagic vesicles in Caenorhabditis elegans, suggesting that this pathway is involved in NM aggregate clearance in higher eukaryotes.Citation79 The fact that NM prions propagate efficiently in mammalian cells argues that a substantial number of prion seeds are either not recognized or escape the cellular clearance machinery.
Sup35 NM Prions Exhibit an Infectious Life Cycle in Mammalian Cells
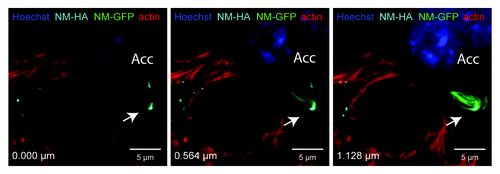
The first evidence for the infectious properties of mammalian cell-derived NM came from experiments using N2a cell extracts containing NM aggregates.Citation35 Addition of this cell extract to N2a cells expressing soluble NM triggered persistent NM aggregation in a subset of cells. Transmission of cytosolic NM aggregates induced the Sup35 prion phenotype also in yeast, arguing that the protein aggregates represent true prions with infectious properties. Remarkably, NM prions in neuroblastoma cells also horizontally infected neighboring cells, in which they induced ongoing prion propagation.Citation41 When N2a cells with NM prions were co-cultured with N2a cells expressing soluble NM, NM aggregation was induced in recipient cells (). The presence of donor NM aggregates in recipient cells strongly argues that direct seed transmission induced the prion phenotype in acceptor cells. The induction efficiency in the recipient cell population ranged between 0.1–0.5% within 24 h. Donor clones producing long fibrillar NM aggregates exhibited lower induction efficiencies compared with clones producing small punctate aggregates. If this was due to the type of aggregate or clonal differences in transmission efficiencies remains to be determined. Cell-to-cell transmission of NM aggregates and subsequent seeding was not only confined to proliferating tumor cell lines, as aggregate induction was also apparent upon coculture of aggregate bearing astrocytes with primary neurons and organotypic slice cultures.Citation41 Thus, NM aggregates gained additional prion characteristics in mammalian cells compared with NM prions present in S. cerevisiae. This spreading behavior of NM prions in tissue culture is reminiscent of mammalian prion dissemination ex vivo.Citation27,Citation80 The horizontal spreading of NM aggregates clearly demonstrates that prion characteristics in mammalian cells are not confined to PrP and that GPI-anchoring is not generally required for prion replication and transmission at least in cell culture models.Citation41
Figure 2. NM-HA aggregates transmitted from N2a donor to acceptor cells (Acc) co-localize with induced NM-GFP aggregates. Confocal microscopy revealed the presence of NM-HA and NM-GFP aggregates in the same N2a cell. Donor cells with induced NM-HA aggregates were co-cultured with acceptor cells expressing soluble NM-GFP. Different layers of a Z-stack are shown (Z distance = 0.564 µm). NM-HA aggregates were stained with anti-HA antibody (light blue) and F-actin was stained with iFluor-546-Phalloidin (red). Nuclei were stained with Hoechst (blue) and NM-GFP is displayed in green. (Scale bar = 5 μm).
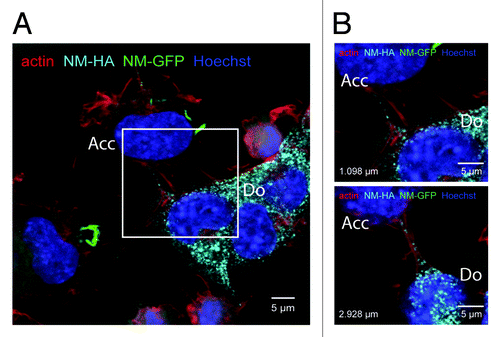
But how do cytosolic NM prions transmit to bystander cells? Possible mechanisms of intercellular aggregate spreading could include an extracellular phase, by transmission via prion secretion or directly by cellular interactions. Aggregates in recipient cells are probably not induced by aggregates released from dying donor cells, since aggregate-bearing cells did not show signs of overt toxicity.Citation41 Indeed, culture medium transferred from aggregate bearing donor cells to recipient cells was significantly less efficient at inducing intracellular NM aggregation than cocultures. NM aggregates spread most efficiently between cells that are in direct contact, suggesting that physical contact between donor and acceptor cells was the prominent route of transmission. Intercellular filopodia-like bridges might be involved in transfer of aggregates, as NM aggregates were sometimes found in actin-rich protrusions connecting donor and acceptor cells (). Interestingly, TNTs also assist in transmission of polyglutamine aggregates between cells, suggesting that intercellular bridges might generally serve as conduits for the exchange of protein aggregates between cells.Citation81 It is also possible that adjacent cells secrete NM aggregates into the intercellular cleft as known for neuronal synapses. In this case, NM prions could be released in the extracellular space, either as naked aggregates or packaged into vesicles, and immediately internalized by the recipient cells. Therefore multiple mechanisms can underlie the aggregate transmission of interconnected cells.
Figure 3. NM-HA aggregates in actin-rich protrusions. (A) Confocal image of a coculture of donor clone 2E (Do) and acceptor cells (Acc) expressing soluble NM-GFP. Cells were fixed after 48 h for immunofluorescence staining. The image shows one layer of a Z-stack. NM-HA aggregates of clone 2E were stained with anti-HA antibody (light blue) and F-actin was stained with iFluor-546-Phalloidin (red). The image shows transmitted light (gray) and NM-GFP in green. Nuclei were stained with Hoechst (blue). NM-GFP in the acceptor cell localizes in inclusions. The white square indicates the inset shown in B. (B) Excerpts of two layers of the Z-stack described above. The distance between the two layers is 1.830 μm. The distances to the bottom are indicated. The images show NM-HA aggregates in actin-rich protrusions between donor and acceptor cell. (Scale bar = 5 μm).
Biological Implications
Our studies on NM prion domain replication have demonstrated that artificial protein aggregates can behave as prions in mammalian cells. So far, the only mammalian protein with clear-cut prion characteristics is PrP. So is the prion propensity unique to PrP or could other mammalian proteins also function as prions? Prions derived from PrP induce uniformly fatal diseases, but prions in lower eukaryotes sometimes fulfill beneficial functions for their hosts. One such example is the (Het-s) prion of the filamentous fungus Podospora anserina required for heterokaryon incompatibility between illegitime mating partners.Citation82 The increasing number of proteins that exhibit prion-like characteristics in lower eukaryotes suggests a functional relevance of prion conformers. It has been proposed that the prion phenotype is an evolutionary conserved mechanism in response to environmental changes.Citation10 Bioinformatics algorithms that analyze compositional similarity with known and synthetic yeast prions have identified several novel prions in yeastCitation83-Citation88 and have predicted candidate proteins in mammals.Citation89 Strikingly, key candidates include proteins known to aggregate in devastating neurodegenerative diseases.Citation89,Citation90 Prion algorithms so far are based on the sequence determinants of yeast prions rich in glutamine (Q) and asparagine (N) and thus will not identify prions that are not enriched in Q and N. Q/N-rich stretches are not a general requirement for prion propensity, and the mammalian PrP,Citation5 the yeast prion Mod5Citation91 and the Het-s prionCitation92 of Podospora anserina are not enriched in Q and N. Refinement of algorithms to include compositional information of novel prions and those without Q/N-rich stretches will likely generate lists of new candidates. A thorough investigation into their prion properties will reveal if those proteins fulfill all prion criteria. In light of the growing number of known prions in lower eukaryotes and the identification of amyloid with biological function also in humans,Citation93,Citation94 it is tempting to speculate that aggregate transfer and conformational templating might be of general biological relevance.
Our study has also important implications for protein misfolding diseases. Increasing evidence argues that intracellular aggregate induction by extracellular proteinaceous seeds is not unique to mammalian and fungal prions, but also a prominent feature of several protein aggregates linked to neurodegenerative diseases in humans.Citation95 In these diseases, unrelated proteins such as Huntingtin, Tau or α-synuclein also template their amyloid states in a seeded-polymerization reaction. Intriguingly, extracellular protein seeds of misfolded, disease-related protein can be taken up by cells and induce an aggregation state of the homologous endogenous protein.Citation49,Citation96-Citation98 Those disease-related cytosolic proteins can even spread within tissues, thereby contributing to pathology progression in mouse models of several neurodegenerative diseases.Citation99-Citation102 The similarities in the seeding and spreading capacities of true prions and other amyloidogenic proteins have blurred boundaries between infectious and non-infectious amyloid. Mammalian prions are true infectious agents, because they (at least several of them) can naturally spread from individual to individual. Epidemiological data do not support an infectious potential of other neurodegenerative diseases. Clearly, there is a lack of fundamental understanding of essential properties of true infectious proteins on all levels, ranging from organismal to cellular to molecular levels.
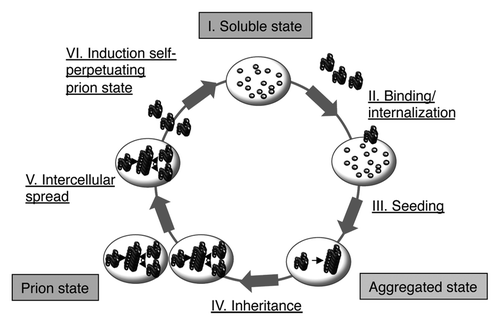
Simple cell culture models such as those discussed here can help to dissect similarities and differences between prions and prion-like proteins on a cellular level. Mitotically active cells are invaluable for studying aggregate multiplication, because induced protein aggregates with low propagation efficiency will be ultimately lost during cell division. The ex vivo life cycles of membrane-bound, PrP-derived prions and cytosolic NM-derived prions are remarkably similar and include six steps: (1) The endogenous protein is in a soluble state, (2) exogenous homotypic seeds can be internalized by cells, (3) the exposure to exogenous seeds causes intracellular aggregate formation, (4) the prion entities are stably inherited during mitosis, (5) aggregates naturally spread and invade neighboring cells, and (6) transmitted aggregates induce the self-perpetuating prion state in the infected cells (). A wealth of data argues that steps 1–3 (invasion of bystander cells and induction of protein aggregation) are shared by many protein aggregates. Oligomers or fibrils formed in vitro from recombinant polyglutamine proteins,Citation97 Tau fragments,Citation50,Citation98,Citation103 mutant superoxide dismutase 1 (SOD1),Citation49 α-synuclein,Citation96,Citation104,Citation105 or patient-derived insoluble TDP-43Citation106 have all been shown to be taken up by cells ex vivo and gain access to the cytosol, where they induce the aggregation of endogenous, homotypic proteins. In some instances, protein aggregates were introduced by transfection, so it is unclear if cells take up those aggregates under more physiological conditions.Citation106
Figure 4. Life cycle of both PrP-derived and NM-derived prions in mammalian cells cultured ex vivo. Note that PrPC and NM differ in their cellular localization. PrPC is a cell surface bound glycoprotein, whereas NM is ectopically expressed in the cytosol. Both proteins are soluble and spontaneous conversion to a prion conformation has not been observed in mammalian cells ex vivo. The prion conformation can be induced by addition of infectious aggregates derived from the respective proteins. For PrP-derived prions, the inoculum is prepared from prion infected tissue or cell culture (for review, see ref. Citation4) or produced in vitro.Citation108 The prion phenotype is faithfully inherited by daughter cells during cell division. For PrP-derived prions, so far unknown susceptibility factors restrict persistent propagation to few cell lines (for review, see ref. Citation4). PrP-derived and NM-derived prions are transmitted horizontally to neighboring cells, in which they induce persistent aggregation of the prion proteins PrP or NM, respectively.
However, efficient secondary nucleation events, natural horizontal transmission, and the induction of self-perpetuating protein aggregation in recipient cells (life cycle steps 4–6) might be efficiently accomplished only by few protein aggregates. Stable inheritance of induced protein aggregates over multiple cell divisions (life cycle step 4) has so far only been reported for mutant SOD1,Citation49 while exogenously induced polyglutamine aggregates were only inefficiently maintained by mitotically active cells.Citation97 The observed differences in aggregate maintenance could be due to different efficiencies in secondary nucleation, aggregate clearance or aggregate toxicity. So far it is unclear if protein aggregates induced by endogenous seeds of Tau fragments, TDP-43 or α-synuclein exhibit mitotic stability. In cellular models, polyglutamine-rich proteins,Citation81 Tau fragments,Citation98,Citation103 α-synucleinCitation101,Citation107 and recombinant SOD1Citation49 have been demonstrated to transmit horizontally from one cell to the next (step 5). In some instances, transmission of those proteins initiated endogenous homotypic protein interactionCitation103 or aggregation.Citation98,Citation101,Citation107 The finding that disease-related, aggregation-prone proteins transmit from one cell to the next (life cycle step 5) has sparked tremendous interest in the intercellular transmission of those disease-related proteins, but it remains to be demonstrated that the transmitted proteins induce a sustained, self-perpetuating aggregation phenotype (life cycle step 6) just as PrP-derived prions (as reviewed in ref. Citation4) or NM-derived prionsCitation41 do. In terms of an infectious entity, the transient induction of a protein aggregate by an exogenous seed would be somewhat reminiscent of an abortive infection by a virus that enters the cell but cannot initiate a full replication cycle that generates infectious progeny. The efficiency of secondary seed generation within the cell is thus a major determinant of prion infectivity. Surprisingly, the necessity for secondary nucleation events that generate secondary nuclei from pre-formed aggregates has so far often been overlooked. The mammalian Sup35 NM model represents a versatile model to study faithful propagation of cytosolic prions on a cellular level.
| Abbreviations: | ||
| GFP | = | green fluorescent protein |
| HA | = | hemagglutinin antibody epitope |
| NM | = | Prion domain of Sup35 |
| PrPC | = | cellular prion protein |
| PrPSc | = | abnormal prion protein |
| TNT | = | tunneling nanotube |
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Sybille Krauss and Walker Jackson for constructive comments on this work. Financial support was provided by the Helmholtz Portfolio Wirkstoffforschung and by the Deutsche Forschungsgemeinschaft (SFB 596 B14, VO 1277/1-2).
References
- Prusiner SB. Novel proteinaceous infectious particles cause scrapie. Science 1982; 216:136 - 44; http://dx.doi.org/10.1126/science.6801762; PMID: 6801762
- Robakis NK, Sawh PR, Wolfe GC, Rubenstein R, Carp RI, Innis MA. Isolation of a cDNA clone encoding the leader peptide of prion protein and expression of the homologous gene in various tissues. Proc Natl Acad Sci U S A 1986; 83:6377 - 81; http://dx.doi.org/10.1073/pnas.83.17.6377; PMID: 3529083
- Brown HR, Goller NL, Rudelli RD, Merz GS, Wolfe GC, Wisniewski HM, Robakis NK. The mRNA encoding the scrapie agent protein is present in a variety of non-neuronal cells. Acta Neuropathol 1990; 80:1 - 6; http://dx.doi.org/10.1007/BF00294214; PMID: 1972856
- Grassmann A, Wolf H, Hofmann J, Graham J, Vorberg I. Cellular aspects of prion replication in vitro. Viruses 2013; 5:374 - 405; http://dx.doi.org/10.3390/v5010374; PMID: 23340381
- Wopfner F, Weidenhöfer G, Schneider R, von Brunn A, Gilch S, Schwarz TF, Werner T, Schätzl HM. Analysis of 27 mammalian and 9 avian PrPs reveals high conservation of flexible regions of the prion protein. J Mol Biol 1999; 289:1163 - 78; http://dx.doi.org/10.1006/jmbi.1999.2831; PMID: 10373359
- Liu JJ, Lindquist S. Oligopeptide-repeat expansions modulate ‘protein-only’ inheritance in yeast. Nature 1999; 400:573 - 6; http://dx.doi.org/10.1038/22919; PMID: 10448860
- Wickner RB, Edskes HK, Roberts BT, Baxa U, Pierce MM, Ross ED, Brachmann A. Prions: proteins as genes and infectious entities. Genes Dev 2004; 18:470 - 85; http://dx.doi.org/10.1101/gad.1177104; PMID: 15037545
- Wickner RB. [URE3] as an altered URE2 protein: evidence for a prion analog in Saccharomyces cerevisiae. Science 1994; 264:566 - 9; http://dx.doi.org/10.1126/science.7909170; PMID: 7909170
- Wickner RB, Taylor KL, Edskes HK, Maddelein ML, Moriyama H, Roberts BT. Prions in Saccharomyces and Podospora spp.: protein-based inheritance. [table of contents.] Microbiol Mol Biol Rev 1999; 63:844 - 61; PMID: 10585968
- Halfmann R, Jarosz DF, Jones SK, Chang A, Lancaster AK, Lindquist S. Prions are a common mechanism for phenotypic inheritance in wild yeasts. Nature 2012; 482:363 - 8; http://dx.doi.org/10.1038/nature10875; PMID: 22337056
- McGlinchey RP, Kryndushkin D, Wickner RB. Suicidal [PSI+] is a lethal yeast prion. Proc Natl Acad Sci U S A 2011; 108:5337 - 41; http://dx.doi.org/10.1073/pnas.1102762108; PMID: 21402947
- Wickner RB, Edskes HK, Bateman D, Kelly AC, Gorkovskiy A. The yeast prions [PSI+] and [URE3] are molecular degenerative diseases. Prion 2011; 5:258 - 62; PMID: 22052353
- Rogoza T, Goginashvili A, Rodionova S, Ivanov M, Viktorovskaya O, Rubel A, Volkov K, Mironova L. Non-Mendelian determinant [ISP+] in yeast is a nuclear-residing prion form of the global transcriptional regulator Sfp1. Proc Natl Acad Sci U S A 2010; 107:10573 - 7; http://dx.doi.org/10.1073/pnas.1005949107; PMID: 20498075
- Orgel LE. Prion replication and secondary nucleation. Chem Biol 1996; 3:413 - 4; http://dx.doi.org/10.1016/S1074-5521(96)90087-3; PMID: 8807870
- Hall D, Edskes H. Silent prions lying in wait: a two-hit model of prion/amyloid formation and infection. J Mol Biol 2004; 336:775 - 86; http://dx.doi.org/10.1016/j.jmb.2003.12.004; PMID: 15095987
- Tanaka M, Collins SR, Toyama BH, Weissman JS. The physical basis of how prion conformations determine strain phenotypes. Nature 2006; 442:585 - 9; http://dx.doi.org/10.1038/nature04922; PMID: 16810177
- Knowles TP, Waudby CA, Devlin GL, Cohen SI, Aguzzi A, Vendruscolo M, Terentjev EM, Welland ME, Dobson CM. An analytical solution to the kinetics of breakable filament assembly. Science 2009; 326:1533 - 7; http://dx.doi.org/10.1126/science.1178250; PMID: 20007899
- Chernoff YO, Lindquist SL, Ono B, Inge-Vechtomov SG, Liebman SW. Role of the chaperone protein Hsp104 in propagation of the yeast prion-like factor [psi+]. [psi+] Science 1995; 268:880 - 4; http://dx.doi.org/10.1126/science.7754373; PMID: 7754373
- Wickner RB, Edskes HK, Bateman DA, Kelly AC, Gorkovskiy A, Dayani Y, Zhou A. Amyloids and yeast prion biology. Biochemistry 2013; 52:1514 - 27; http://dx.doi.org/10.1021/bi301686a; PMID: 23379365
- Winkler J, Tyedmers J, Bukau B, Mogk A. Chaperone networks in protein disaggregation and prion propagation. J Struct Biol 2012; 179:152 - 60; http://dx.doi.org/10.1016/j.jsb.2012.05.002; PMID: 22580344
- Derkatch IL, Bradley ME, Zhou P, Chernoff YO, Liebman SW. Genetic and environmental factors affecting the de novo appearance of the [PSI+] prion in Saccharomyces cerevisiae. Genetics 1997; 147:507 - 19; PMID: 9335589
- Derkatch IL, Bradley ME, Hong JY, Liebman SW. Prions affect the appearance of other prions: the story of [PIN(+)]. Cell 2001; 106:171 - 82; http://dx.doi.org/10.1016/S0092-8674(01)00427-5; PMID: 11511345
- Osherovich LZ, Weissman JS. Multiple Gln/Asn-rich prion domains confer susceptibility to induction of the yeast [PSI(+)] prion. Cell 2001; 106:183 - 94; http://dx.doi.org/10.1016/S0092-8674(01)00440-8; PMID: 11511346
- Uptain SM, Lindquist S. Prions as protein-based genetic elements. Annu Rev Microbiol 2002; 56:703 - 41; http://dx.doi.org/10.1146/annurev.micro.56.013002.100603; PMID: 12142498
- Wickner RB, Fujimura T, Esteban R. Viruses and prions of Saccharomyces cerevisiae. Adv Virus Res 2013; 86:1 - 36; http://dx.doi.org/10.1016/B978-0-12-394315-6.00001-5; PMID: 23498901
- Aguzzi A. Prions and the immune system: a journey through gut, spleen, and nerves. Adv Immunol 2003; 81:123 - 71; http://dx.doi.org/10.1016/S0065-2776(03)81004-0; PMID: 14711055
- Kanu N, Imokawa Y, Drechsel DN, Williamson RA, Birkett CR, Bostock CJ, Brockes JP. Transfer of scrapie prion infectivity by cell contact in culture. Curr Biol 2002; 12:523 - 30; http://dx.doi.org/10.1016/S0960-9822(02)00722-4; PMID: 11937020
- Gousset K, Schiff E, Langevin C, Marijanovic Z, Caputo A, Browman DT, Chenouard N, de Chaumont F, Martino A, Enninga J, et al. Prions hijack tunnelling nanotubes for intercellular spread. Nat Cell Biol 2009; 11:328 - 36; http://dx.doi.org/10.1038/ncb1841; PMID: 19198598
- Gerdes HH, Carvalho RN. Intercellular transfer mediated by tunneling nanotubes. Curr Opin Cell Biol 2008; 20:470 - 5; http://dx.doi.org/10.1016/j.ceb.2008.03.005; PMID: 18456488
- Leblanc P, Alais S, Porto-Carreiro I, Lehmann S, Grassi J, Raposo G, Darlix JL. Retrovirus infection strongly enhances scrapie infectivity release in cell culture. EMBO J 2006; 25:2674 - 85; http://dx.doi.org/10.1038/sj.emboj.7601162; PMID: 16724107
- Vella LJ, Sharples RA, Lawson VA, Masters CL, Cappai R, Hill AF. Packaging of prions into exosomes is associated with a novel pathway of PrP processing. J Pathol 2007; 211:582 - 90; http://dx.doi.org/10.1002/path.2145; PMID: 17334982
- Alais S, Simoes S, Baas D, Lehmann S, Raposo G, Darlix JL, Leblanc P. Mouse neuroblastoma cells release prion infectivity associated with exosomal vesicles. Biol Cell 2008; 100:603 - 15; http://dx.doi.org/10.1042/BC20080025; PMID: 18422484
- Castro-Seoane R, Hummerich H, Sweeting T, Tattum MH, Linehan JM, Fernandez de Marco M, Brandner S, Collinge J, Klöhn PC. Plasmacytoid dendritic cells sequester high prion titres at early stages of prion infection. PLoS Pathog 2012; 8:e1002538; http://dx.doi.org/10.1371/journal.ppat.1002538; PMID: 22359509
- Caughey B, Baron GS, Chesebro B, Jeffrey M. Getting a grip on prions: oligomers, amyloids, and pathological membrane interactions. Annu Rev Biochem 2009; 78:177 - 204; http://dx.doi.org/10.1146/annurev.biochem.78.082907.145410; PMID: 19231987
- Krammer C, Kryndushkin D, Suhre MH, Kremmer E, Hofmann A, Pfeifer A, Scheibel T, Wickner RB, Schätzl HM, Vorberg I. The yeast Sup35NM domain propagates as a prion in mammalian cells. Proc Natl Acad Sci U S A 2009; 106:462 - 7; http://dx.doi.org/10.1073/pnas.0811571106; PMID: 19114662
- Krammer C, Schätzl HM, Vorberg I. Prion-like propagation of cytosolic protein aggregates: insights from cell culture models. Prion 2009; 3:206 - 12; http://dx.doi.org/10.4161/pri.3.4.10013; PMID: 19901539
- Doel SM, McCready SJ, Nierras CR, Cox BS. The dominant PNM2- mutation which eliminates the psi factor of Saccharomyces cerevisiae is the result of a missense mutation in the SUP35 gene. Genetics 1994; 137:659 - 70; PMID: 8088511
- Ter-Avanesyan MD, Dagkesamanskaya AR, Kushnirov VV, Smirnov VN. The SUP35 omnipotent suppressor gene is involved in the maintenance of the non-Mendelian determinant [psi+] in the yeast Saccharomyces cerevisiae. Genetics 1994; 137:671 - 6; PMID: 8088512
- Derkatch IL, Chernoff YO, Kushnirov VV, Inge-Vechtomov SG, Liebman SW. Genesis and variability of [PSI] prion factors in Saccharomyces cerevisiae. Genetics 1996; 144:1375 - 86; PMID: 8978027
- Liu JJ, Sondheimer N, Lindquist SL. Changes in the middle region of Sup35 profoundly alter the nature of epigenetic inheritance for the yeast prion [PSI+]. [PSI+] Proc Natl Acad Sci U S A 2002; 99:Suppl 4 16446 - 53; http://dx.doi.org/10.1073/pnas.252652099; PMID: 12461168
- Hofmann JP, Denner P, Nussbaum-Krammer C, Kuhn PH, Suhre MH, Scheibel T, Lichtenthaler SF, Schätzl HM, Bano D, Vorberg IM. Cell-to-cell propagation of infectious cytosolic protein aggregates. Proc Natl Acad Sci U S A 2013; 110:5951 - 6; http://dx.doi.org/10.1073/pnas.1217321110; PMID: 23509289
- Sparrer HE, Santoso A, Szoka FC Jr., Weissman JS. Evidence for the prion hypothesis: induction of the yeast [PSI+] factor by in vitro- converted Sup35 protein. Science 2000; 289:595 - 9; http://dx.doi.org/10.1126/science.289.5479.595; PMID: 10915616
- Tanaka M, Chien P, Naber N, Cooke R, Weissman JS. Conformational variations in an infectious protein determine prion strain differences. Nature 2004; 428:323 - 8; http://dx.doi.org/10.1038/nature02392; PMID: 15029196
- Brachmann A, Baxa U, Wickner RB. Prion generation in vitro: amyloid of Ure2p is infectious. EMBO J 2005; 24:3082 - 92; http://dx.doi.org/10.1038/sj.emboj.7600772; PMID: 16096644
- King CY, Diaz-Avalos R. Protein-only transmission of three yeast prion strains. Nature 2004; 428:319 - 23; http://dx.doi.org/10.1038/nature02391; PMID: 15029195
- Gauczynski S, Nikles D, El-Gogo S, Papy-Garcia D, Rey C, Alban S, Barritault D, Lasmezas CI, Weiss S. The 37-kDa/67-kDa laminin receptor acts as a receptor for infectious prions and is inhibited by polysulfated glycanes. J Infect Dis 2006; 194:702 - 9; http://dx.doi.org/10.1086/505914; PMID: 16897671
- Wadia JS, Schaller M, Williamson RA, Dowdy SF. Pathologic prion protein infects cells by lipid-raft dependent macropinocytosis. PLoS One 2008; 3:e3314; http://dx.doi.org/10.1371/journal.pone.0003314; PMID: 19390657
- Jen A, Parkyn CJ, Mootoosamy RC, Ford MJ, Warley A, Liu Q, Bu G, Baskakov IV, Moestrup S, McGuinness L, et al. Neuronal low-density lipoprotein receptor-related protein 1 binds and endocytoses prion fibrils via receptor cluster 4. J Cell Sci 2010; 123:246 - 55; http://dx.doi.org/10.1242/jcs.058099; PMID: 20048341
- Münch C, O’Brien J, Bertolotti A. Prion-like propagation of mutant superoxide dismutase-1 misfolding in neuronal cells. Proc Natl Acad Sci U S A 2011; 108:3548 - 53; http://dx.doi.org/10.1073/pnas.1017275108; PMID: 21321227
- Wu JW, Herman M, Liu L, Simoes S, Acker CM, Figueroa H, Steinberg JI, Margittai M, Kayed R, Zurzolo C, et al. Small misfolded Tau species are internalized via bulk endocytosis and anterogradely and retrogradely transported in neurons. J Biol Chem 2013; 288:1856 - 70; http://dx.doi.org/10.1074/jbc.M112.394528; PMID: 23188818
- Holmes BB, Devos SL, Kfoury N, Li M, Jacks R, Yanamandra K, Ouidja MO, Brodsky FM, Marasa J, Bagchi DP, et al. Heparan sulfate proteoglycans mediate internalization and propagation of specific proteopathic seeds. Proc Natl Acad Sci U S A 2013; 110:E3138 - 47; http://dx.doi.org/10.1073/pnas.1301440110; PMID: 23898162
- Bucciantini M, Nosi D, Forzan M, Russo E, Calamai M, Pieri L, Formigli L, Quercioli F, Soria S, Pavone F, et al. Toxic effects of amyloid fibrils on cell membranes: the importance of ganglioside GM1. FASEB J 2012; 26:818 - 31; http://dx.doi.org/10.1096/fj.11-189381; PMID: 22071505
- Race RE, Fadness LH, Chesebro B. Characterization of scrapie infection in mouse neuroblastoma cells. J Gen Virol 1987; 68:1391 - 9; http://dx.doi.org/10.1099/0022-1317-68-5-1391; PMID: 3106566
- Butler DA, Scott MR, Bockman JM, Borchelt DR, Taraboulos A, Hsiao KK, Kingsbury DT, Prusiner SB. Scrapie-infected murine neuroblastoma cells produce protease-resistant prion proteins. J Virol 1988; 62:1558 - 64; PMID: 3282080
- Vorberg I, Raines A, Story B, Priola SA. Susceptibility of common fibroblast cell lines to transmissible spongiform encephalopathy agents. J Infect Dis 2004; 189:431 - 9; http://dx.doi.org/10.1086/381166; PMID: 14745700
- Ghaemmaghami S, Phuan PW, Perkins B, Ullman J, May BC, Cohen FE, Prusiner SB. Cell division modulates prion accumulation in cultured cells. Proc Natl Acad Sci U S A 2007; 104:17971 - 6; http://dx.doi.org/10.1073/pnas.0708372104; PMID: 17989223
- Aguilaniu H, Gustafsson L, Rigoulet M, Nyström T. Asymmetric inheritance of oxidatively damaged proteins during cytokinesis. Science 2003; 299:1751 - 3; http://dx.doi.org/10.1126/science.1080418; PMID: 12610228
- Rujano MA, Bosveld F, Salomons FA, Dijk F, van Waarde MA, van der Want JJ, de Vos RA, Brunt ER, Sibon OC, Kampinga HH. Polarised asymmetric inheritance of accumulated protein damage in higher eukaryotes. PLoS Biol 2006; 4:e417; http://dx.doi.org/10.1371/journal.pbio.0040417; PMID: 17147470
- Parsell DA, Kowal AS, Singer MA, Lindquist S. Protein disaggregation mediated by heat-shock protein Hsp104. Nature 1994; 372:475 - 8; http://dx.doi.org/10.1038/372475a0; PMID: 7984243
- Weibezahn J, Bukau B, Mogk A. Unscrambling an egg: protein disaggregation by AAA+ proteins. Microb Cell Fact 2004; 3:1; http://dx.doi.org/10.1186/1475-2859-3-1; PMID: 14728719
- Chernoff YO, Newnam GP, Kumar J, Allen K, Zink AD. Evidence for a protein mutator in yeast: role of the Hsp70-related chaperone ssb in formation, stability, and toxicity of the [PSI] prion. Mol Cell Biol 1999; 19:8103 - 12; PMID: 10567536
- Kushnirov VV, Kryndushkin DS, Boguta M, Smirnov VN, Ter-Avanesyan MD. Chaperones that cure yeast artificial [PSI+] and their prion-specific effects. Curr Biol 2000; 10:1443 - 6; http://dx.doi.org/10.1016/S0960-9822(00)00802-2; PMID: 11102806
- Cox B, Ness F, Tuite M. Analysis of the generation and segregation of propagons: entities that propagate the [PSI+] prion in yeast. Genetics 2003; 165:23 - 33; PMID: 14504215
- Kryndushkin DS, Engel A, Edskes H, Wickner RB. Molecular chaperone Hsp104 can promote yeast prion generation. Genetics 2011; 188:339 - 48; http://dx.doi.org/10.1534/genetics.111.127779; PMID: 21467567
- Wickner RB. Discovering protein-based inheritance through yeast genetics. J Biol Chem 2012; 287:14432 - 42; http://dx.doi.org/10.1074/jbc.X112.355636; PMID: 22396539
- Castilla J, Saá P, Hetz C, Soto C. In vitro generation of infectious scrapie prions. Cell 2005; 121:195 - 206; http://dx.doi.org/10.1016/j.cell.2005.02.011; PMID: 15851027
- Saá P, Castilla J, Soto C. Ultra-efficient replication of infectious prions by automated protein misfolding cyclic amplification. J Biol Chem 2006; 281:35245 - 52; http://dx.doi.org/10.1074/jbc.M603964200; PMID: 16982620
- Newnam GP, Wegrzyn RD, Lindquist SL, Chernoff YO. Antagonistic interactions between yeast chaperones Hsp104 and Hsp70 in prion curing. Mol Cell Biol 1999; 19:1325 - 33; PMID: 9891066
- Murray AN, Solomon JP, Wang YJ, Balch WE, Kelly JW. Discovery and characterization of a mammalian amyloid disaggregation activity. Protein Sci 2010; 19:836 - 46; http://dx.doi.org/10.1002/pro.363; PMID: 20162625
- Shorter J. The mammalian disaggregase machinery: Hsp110 synergizes with Hsp70 and Hsp40 to catalyze protein disaggregation and reactivation in a cell-free system. PLoS One 2011; 6:e26319; http://dx.doi.org/10.1371/journal.pone.0026319; PMID: 22022600
- Rampelt H, Kirstein-Miles J, Nillegoda NB, Chi K, Scholz SR, Morimoto RI, Bukau B. Metazoan Hsp70 machines use Hsp110 to power protein disaggregation. EMBO J 2012; 31:4221 - 35; http://dx.doi.org/10.1038/emboj.2012.264; PMID: 22990239
- Raviol H, Sadlish H, Rodriguez F, Mayer MP, Bukau B. Chaperone network in the yeast cytosol: Hsp110 is revealed as an Hsp70 nucleotide exchange factor. EMBO J 2006; 25:2510 - 8; http://dx.doi.org/10.1038/sj.emboj.7601139; PMID: 16688211
- Dragovic Z, Broadley SA, Shomura Y, Bracher A, Hartl FU. Molecular chaperones of the Hsp110 family act as nucleotide exchange factors of Hsp70s. EMBO J 2006; 25:2519 - 28; http://dx.doi.org/10.1038/sj.emboj.7601138; PMID: 16688212
- Yasuda K, Nakai A, Hatayama T, Nagata K. Cloning and expression of murine high molecular mass heat shock proteins, HSP105. J Biol Chem 1995; 270:29718 - 23; http://dx.doi.org/10.1074/jbc.270.50.29718; PMID: 8530361
- Vos MJ, Hageman J, Carra S, Kampinga HH. Structural and functional diversities between members of the human HSPB, HSPH, HSPA, and DNAJ chaperone families. Biochemistry 2008; 47:7001 - 11; http://dx.doi.org/10.1021/bi800639z; PMID: 18557634
- Duennwald ML, Echeverria A, Shorter J. Small heat shock proteins potentiate amyloid dissolution by protein disaggregases from yeast and humans. PLoS Biol 2012; 10:e1001346; http://dx.doi.org/10.1371/journal.pbio.1001346; PMID: 22723742
- Aguib Y, Heiseke A, Gilch S, Riemer C, Baier M, Schätzl HM, Ertmer A. Autophagy induction by trehalose counteracts cellular prion infection. Autophagy 2009; 5:361 - 9; http://dx.doi.org/10.4161/auto.5.3.7662; PMID: 19182537
- Heiseke A, Aguib Y, Riemer C, Baier M, Schätzl HM. Lithium induces clearance of protease resistant prion protein in prion-infected cells by induction of autophagy. J Neurochem 2009; 109:25 - 34; http://dx.doi.org/10.1111/j.1471-4159.2009.05906.x; PMID: 19183256
- Nussbaum-Krammer CI, Park KW, Li L, Melki R, Morimoto RI. Spreading of a prion domain from cell-to-cell by vesicular transport in Caenorhabditis elegans. PLoS Genet 2013; 9:e1003351; http://dx.doi.org/10.1371/journal.pgen.1003351; PMID: 23555277
- Paquet S, Langevin C, Chapuis J, Jackson GS, Laude H, Vilette D. Efficient dissemination of prions through preferential transmission to nearby cells. J Gen Virol 2007; 88:706 - 13; http://dx.doi.org/10.1099/vir.0.82336-0; PMID: 17251590
- Costanzo M, Abounit S, Marzo L, Danckaert A, Chamoun Z, Roux P, Zurzolo C. Transfer of polyglutamine aggregates in neuronal cells occurs in tunneling nanotubes. J Cell Sci 2013; 126:3678 - 85; http://dx.doi.org/10.1242/jcs.126086; PMID: 23781027
- Saupe SJ. The [Het-s] prion of Podospora anserina and its role in heterokaryon incompatibility. Semin Cell Dev Biol 2011; 22:460 - 8; http://dx.doi.org/10.1016/j.semcdb.2011.02.019; PMID: 21334447
- Michelitsch MD, Weissman JS. A census of glutamine/asparagine-rich regions: implications for their conserved function and the prediction of novel prions. Proc Natl Acad Sci U S A 2000; 97:11910 - 5; http://dx.doi.org/10.1073/pnas.97.22.11910; PMID: 11050225
- Lashuel HA, Pappu RV. Amyloids go genomic: insights regarding the sequence determinants of prion formation from genome-wide studies. Chembiochem 2009; 10:1951 - 4; http://dx.doi.org/10.1002/cbic.200900373; PMID: 19598186
- Alberti S, Halfmann R, King O, Kapila A, Lindquist S. A systematic survey identifies prions and illuminates sequence features of prionogenic proteins. Cell 2009; 137:146 - 58; http://dx.doi.org/10.1016/j.cell.2009.02.044; PMID: 19345193
- Toombs JA, McCarty BR, Ross ED. Compositional determinants of prion formation in yeast. Mol Cell Biol 2010; 30:319 - 32; http://dx.doi.org/10.1128/MCB.01140-09; PMID: 19884345
- Halfmann R, Alberti S, Krishnan R, Lyle N, O’Donnell CW, King OD, Berger B, Pappu RV, Lindquist S. Opposing effects of glutamine and asparagine govern prion formation by intrinsically disordered proteins. Mol Cell 2011; 43:72 - 84; http://dx.doi.org/10.1016/j.molcel.2011.05.013; PMID: 21726811
- Toombs JA, Petri M, Paul KR, Kan GY, Ben-Hur A, Ross ED. De novo design of synthetic prion domains. Proc Natl Acad Sci U S A 2012; 109:6519 - 24; http://dx.doi.org/10.1073/pnas.1119366109; PMID: 22474356
- King OD, Gitler AD, Shorter J. The tip of the iceberg: RNA-binding proteins with prion-like domains in neurodegenerative disease. Brain Res 2012; 1462:61 - 80; http://dx.doi.org/10.1016/j.brainres.2012.01.016; PMID: 22445064
- Kim HJ, Kim NC, Wang YD, Scarborough EA, Moore J, Diaz Z, MacLea KS, Freibaum B, Li S, Molliex A, et al. Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature 2013; 495:467 - 73; http://dx.doi.org/10.1038/nature11922; PMID: 23455423
- Suzuki G, Shimazu N, Tanaka M. A yeast prion, Mod5, promotes acquired drug resistance and cell survival under environmental stress. Science 2012; 336:355 - 9; http://dx.doi.org/10.1126/science.1219491; PMID: 22517861
- Coustou V, Deleu C, Saupe S, Begueret J. The protein product of the het-s heterokaryon incompatibility gene of the fungus Podospora anserina behaves as a prion analog. Proc Natl Acad Sci U S A 1997; 94:9773 - 8; http://dx.doi.org/10.1073/pnas.94.18.9773; PMID: 9275200
- Fowler DM, Koulov AV, Alory-Jost C, Marks MS, Balch WE, Kelly JW. Functional amyloid formation within mammalian tissue. PLoS Biol 2006; 4:e6; http://dx.doi.org/10.1371/journal.pbio.0040006; PMID: 16300414
- Maji SK, Perrin MH, Sawaya MR, Jessberger S, Vadodaria K, Rissman RA, Singru PS, Nilsson KP, Simon R, Schubert D, et al. Functional amyloids as natural storage of peptide hormones in pituitary secretory granules. Science 2009; 325:328 - 32; http://dx.doi.org/10.1126/science.1173155; PMID: 19541956
- Goedert M, Clavaguera F, Tolnay M. The propagation of prion-like protein inclusions in neurodegenerative diseases. Trends Neurosci 2010; 33:317 - 25; http://dx.doi.org/10.1016/j.tins.2010.04.003; PMID: 20493564
- Danzer KM, Krebs SK, Wolff M, Birk G, Hengerer B. Seeding induced by alpha-synuclein oligomers provides evidence for spreading of alpha-synuclein pathology. J Neurochem 2009; 111:192 - 203; http://dx.doi.org/10.1111/j.1471-4159.2009.06324.x; PMID: 19686384
- Ren PH, Lauckner JE, Kachirskaia I, Heuser JE, Melki R, Kopito RR. Cytoplasmic penetration and persistent infection of mammalian cells by polyglutamine aggregates. Nat Cell Biol 2009; 11:219 - 25; http://dx.doi.org/10.1038/ncb1830; PMID: 19151706
- Frost B, Jacks RL, Diamond MI. Propagation of tau misfolding from the outside to the inside of a cell. J Biol Chem 2009; 284:12845 - 52; http://dx.doi.org/10.1074/jbc.M808759200; PMID: 19282288
- Guo JL, Lee VM. Seeding of normal Tau by pathological Tau conformers drives pathogenesis of Alzheimer-like tangles. J Biol Chem 2011; 286:15317 - 31; http://dx.doi.org/10.1074/jbc.M110.209296; PMID: 21372138
- Luk KC, Kehm VM, Zhang B, O’Brien P, Trojanowski JQ, Lee VM. Intracerebral inoculation of pathological α-synuclein initiates a rapidly progressive neurodegenerative α-synucleinopathy in mice. J Exp Med 2012; 209:975 - 86; http://dx.doi.org/10.1084/jem.20112457; PMID: 22508839
- Desplats P, Lee HJ, Bae EJ, Patrick C, Rockenstein E, Crews L, Spencer B, Masliah E, Lee SJ. Inclusion formation and neuronal cell death through neuron-to-neuron transmission of alpha-synuclein. Proc Natl Acad Sci U S A 2009; 106:13010 - 5; http://dx.doi.org/10.1073/pnas.0903691106; PMID: 19651612
- Clavaguera F, Bolmont T, Crowther RA, Abramowski D, Frank S, Probst A, Fraser G, Stalder AK, Beibel M, Staufenbiel M, et al. Transmission and spreading of tauopathy in transgenic mouse brain. Nat Cell Biol 2009; 11:909 - 13; http://dx.doi.org/10.1038/ncb1901; PMID: 19503072
- Kfoury N, Holmes BB, Jiang H, Holtzman DM, Diamond MI. Trans-cellular propagation of Tau aggregation by fibrillar species. J Biol Chem 2012; 287:19440 - 51; http://dx.doi.org/10.1074/jbc.M112.346072; PMID: 22461630
- Luk KC, Song C, O’Brien P, Stieber A, Branch JR, Brunden KR, Trojanowski JQ, Lee VM. Exogenous alpha-synuclein fibrils seed the formation of Lewy body-like intracellular inclusions in cultured cells. Proc Natl Acad Sci U S A 2009; 106:20051 - 6; PMID: 19892735
- Volpicelli-Daley LA, Luk KC, Patel TP, Tanik SA, Riddle DM, Stieber A, Meaney DF, Trojanowski JQ, Lee VM. Exogenous α-synuclein fibrils induce Lewy body pathology leading to synaptic dysfunction and neuron death. Neuron 2011; 72:57 - 71; http://dx.doi.org/10.1016/j.neuron.2011.08.033; PMID: 21982369
- Nonaka T, Masuda-Suzukake M, Arai T, Hasegawa Y, Akatsu H, Obi T, Yoshida M, Murayama S, Mann DM, Akiyama H, et al. Prion-like properties of pathological TDP-43 aggregates from diseased brains. Cell Rep 2013; 4:124 - 34; http://dx.doi.org/10.1016/j.celrep.2013.06.007; PMID: 23831027
- Lee HJ, Suk JE, Patrick C, Bae EJ, Cho JH, Rho S, Hwang D, Masliah E, Lee SJ. Direct transfer of alpha-synuclein from neuron to astroglia causes inflammatory responses in synucleinopathies. J Biol Chem 2010; 285:9262 - 72; http://dx.doi.org/10.1074/jbc.M109.081125; PMID: 20071342
- Wang F, Wang X, Yuan CG, Ma J. Generating a prion with bacterially expressed recombinant prion protein. Science 2010; 327:1132 - 5; http://dx.doi.org/10.1126/science.1183748; PMID: 20110469