Abstract
Creutzfeldt-Jakob disease (CJD) is a heterogenic neurodegenerative disorder associated with abnormal post-translational processing of cellular prion protein (PrPc). CJD displays distinctive clinical and pathological features which correlate with the genotype at the codon 129 (methionine or valine: M or V respectively) in the prion protein gene and with size of the protease-resistant core of the abnormal prion protein PrPsc (type 1: 20/21 kDa and type 2: 19 kDa). MM1 and VV2 are the most common sporadic CJD (sCJD) subtypes. PrP mRNA expression levels in the frontal cortex and cerebellum are reduced in sCJD in a form subtype-dependent. Total PrP protein levels and PrPsc levels in the frontal cortex and cerebellum accumulate differentially in sCJD MM1 and sCJD VV2 with no relation between PrPsc deposition and spongiform degeneration and neuron loss, but with microgliosis, and IL6 and TNF-α response. In the CSF, reduced PrPc, the only form present in this compartment, occurs in sCJD MM1 and VV2. PrP mRNA expression is also reduced in the frontal cortex in advanced stages of Alzheimer disease, Lewy body disease, progressive supranuclear palsy, and frontotemporal lobe degeneration, but PrPc levels in brain varies from one disease to another. Reduced PrPc levels in CSF correlate with PrP mRNA expression in brain, which in turn reflects severity of degeneration in sCJD.
Introduction
Prion diseases are currently untreatable neurodegenerative disorders, united by a common molecular core. The normal prion protein (PrPc), rich in α-helical structure, undergoes a conformational change turning into the abnormal disease-related form (PrPsc) comprised predominately by β-sheets.Citation1
PrPsc acts as the infectious agent of the disease and forms PrP aggregates in the brain which are eventually associated with neurodegeneration. Although the in vivo pathway for this conversion is still unknown, it has been proposed that PrPsc has the ability to self-perpetuate its structure and thus, upon binding to PrPc, promotes the conformational shift of the physiological form into the pathogenic form.Citation2
The most common prion disease in humans is Creutzfeldt-Jakob disease (CJD), mostly affecting the middle-aged and elderly people. CJD may occur as sporadic (sCJD), which accounts for the majority of CJD cases (85%), familial (fCJD), or infectious disease (iCJD). The basic neuropathology associated with CJD is characterized by spongiform degeneration, neuronal loss, synaptic loss, astrogliosis, microglial activation and accumulation of PrPsc in the brain.Citation3 However, early symptoms, clinical manifestations, disease duration and particularities of neuropathological findings largely depend on the composition of codon 129 (methionione or valine) and PrP type. PrPsc typing is based on the protease digestion pattern and the molecular weight of the lower PrPsc band of 20/21 kDa or 19 kDa, respectively for type 1 and type 2, as revealed by gel electrophoresis and western blotting of brain homogenates.Citation4,Citation5 The most common subtypes varieties of sCJD are MM1 and VV2 accounting for 57% and 14% of sCJD cases, respectively.Citation6-Citation8
In the central nervous system, PrPc is mainly localized in synaptic buttons and synapses.Citation9 PrPc is mandatory for prion propagation as PrP-null mice are resistant to prion diseases.Citation10 However, little is known about the modulation of PrPc expression in distinct neurodegenerative settings, including sCJD, and the correlation between PrP mRNA, PrPc and PrPsc in sCJD.
These aspects are important as they not only improve our knowledge on the mechanistic aspects of these diseases, but also help understand the rationale for the use of certain molecules as biomarkers of neurodegenerative diseases.
Proteomic studies performed in CSF are a valuable tool in the search for new potential CJD biomarkers.Citation11-Citation14 PrPc is present in the CSF under physiological conditions,Citation15 and levels are reduced in the CSF of sCJD patients.Citation16,Citation17 It has been discussed whether these findings are related to unspecific brain degeneration and neuronal loss or to specific disease-related pathophysiology.
The aims of the present study are to: (1) characterize the correlation between PrP mRNA and PrP protein (PrPc and PrPsc) in frontal cortex and cerebellum of sCJD MM1 and sCJD VV2; (2) analyze possible correlates between PrP mRNA and PrPsc with neuropathological hallmarks of sCJD; (3) compare these features with the recent observations of reduced PrPc levels in the CSF in sCJD,Citation16 and (4) compare the same PrP-related parameters in sCJD and other neurodegenerative diseases manifested by cognitive impairment and dementia including Alzheimer disease (AD), Lewy body disease (LBD), progressive supranuclear palsy (PSP), and frontotemporal lobar degeneration (FTLD) due to mutations in the tau gene (fFTLD-tau) and FTLD-TDP-43.
Results
Prion protein mRNA and protein expression in sCJD MM1 and VV2 subtypes
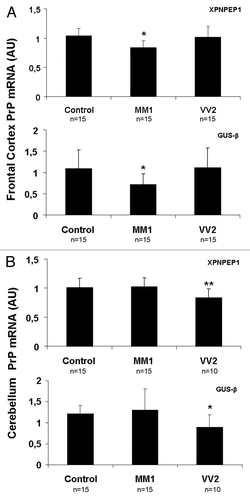
PrP mRNA expression was assessed by RT-qPCR in frontal cortex and cerebellum from sCJD cases with subtypes MM1 and VV2, and age-matched controls. A commercially available PrP probe and two different housekeeping genes were used to avoid normalization bias. A significant decrease in PrP mRNA levels was observed in frontal cortex of MM1 cases () and in cerebellum of VV2 cases () when compared with their corresponding controls. Similar results were obtained with the two housekeeping genes used for normalization.
Figure 1. PrP mRNA in sCJD brain samples. RNA average expression levels of PrP in the frontal cortex (A) and cerebellum (B) of control, sCJD MM1 and sCJD VV2 subtypes, as determined by TaqMan PCR assays. Values for PrP mRNA are normalized using XPNPEP1 (A and B, upper panels) and GUSβ (A and B lower panels) as internal controls (housekeeping genes). Reduced PrP mRNA is seen in the frontal cortex in sCJD MM1 and cerebellum in sCJD VV2. Similar results are obtained with the housekeeping genes. *P > 0.05; **P > 0.01, control vs sCJD. AU: arbitrary units.
The same samples were then analyzed by western blot. Total homogenates of the frontal cortex and cerebellum treated with proteinase K (PK) revealed the typical patterns of upper glycosylated bands and a lower glycosylated band of 20/21 kDa and 19 kDa, respectively for PrP1 and PrP2, whereas no PK-resistant PrP was found in the frontal cortex and cerebellum in control cases (Fig. S1A).
Total PrP protein, obtained without PK treatment, was analyzed in the same homogenates in frontal cortex and cerebellum (). PNGase treatment of control, sCJD MM1 and VV2 confirmed that the low molecular band in sCJD samples corresponded to un-glycosylated PrP forms (Fig. S1B).
Figure 2. PrP protein expression in sCJD brain and CSF samples. (A) western blot analysis of PrP using the PrP antibody 3F4 in the frontal cortex of control, sCJD MM1 and sCJD VV2 cases represented by five cases per condition and using β-actin immunostaining and Coomassie Blue to normalize total protein loading are shown in the upper panel. Densiometric values of all the cases analyzed by western blot: control (n = 15), sCJD MM1 (n = 15), sCJD VV2 (n = 15) show significant decrease of total PrP in MM1 cases (lower panel). (B) western blot analysis of PrP using the PrP antibody 3F4 in the cerebellum of control, sCJD MM1 and sCJD VV2 cases represented by five cases per condition are shown in the upper panel. β-actin immunostaining and Coomassie-Blue were used to normalize total protein loaded into the gel. Densiometric assessment of all the cases analyzed by western blot: control (n = 15), sCJD MM1 (n = 15), sCJD VV2 (n = 14) reveals a marked increase in PrP protein expression levels in the cerebellum of VV2 (lower panel). Note the presence of the lower band of 20 kDa or 19 kDa only in sCJD cases. (C) ELISA analysis using Platelia BSE-Detection Kit (Bio-Rad Laboratories GmbH) of PrPc levels in the CSF of control (Con), sCJD MM1 and sCJD VV2. *P > 0.05; *P > 0.005; ***P > 0.001: control vs sCJD, ###P > 0.001: sCJD MM1 vs VV2. AU: arbitrary units.
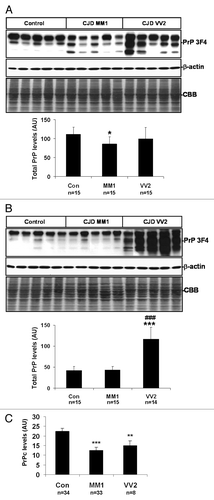
Densitometric analysis revealed a slight but significant decrease in total PrP protein expression levels in the frontal cortex in sCJD MM1, but not in sCJD VV2, when compared with control samples. This was accompanied by an increase in the expression levels of the un-glycosylated, lower molecular weight band in sCJD MM1 and sCJD VV2 samples (; Fig. S2A and B). Regarding the cerebellum, no differences were seen between controls and sCJD MM1 cases in the total PrP protein and in the amount of the un-glycosylated bands. However, a marked increase in total PrP protein together with an increase in the amount of the un-glycosylated bands was found in sCJD VV2 (, Fig. S2C and D).
PrP expression levels were also analyzed by means of ELISA in the CSF of controls and sCJD. A significant reduction in PrPc levels was detected in sCJD MM1 and VV2 without significant differences between subtypes ().
PrPsc deposits in sCJD MM1 and VV2 subtypes
Immunohistochemistry analysis of PK-treated slices with anti-PrP antibodies (clone 3F4) was performed for both subtypes and brain regions. PrPsc deposition with a predominant synaptic-like pattern was observed in frontal cortex and cerebellum in sCJD MM1 subtype. A synaptic-like pattern together with occasional small plaques and perineuronal PrP deposits occurred in the frontal cortex of sCJD VV2, whereas marked PrP plaque-like deposition was observed mainly in the granular and Purkinje cell layers, and white matter of the cerebellum in sCJD VV2 (; ).
Figure 3. PrPsc deposition in sCJD brain samples. PrPsc immunohistochemistry shows differential PrP deposition in frontal cortex and cerebellum in sCJD MM1 and sCJD VV2 cases. Sections were pre-incubated with PK prior to PrPsc immunohistochemistry. Synaptic pattern of PrPsc deposition in the frontal cortex and molecular layer of the cerebellum characterizes MM1, whereas synaptic and plaque-like PrPsc deposition, mainly in the cerebellum, characterizes VV2.
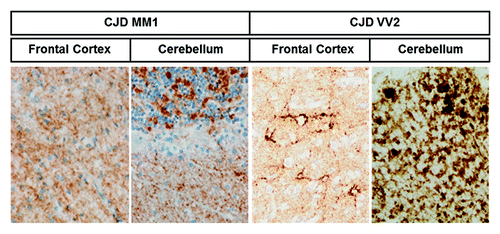
Table 1. Summary of the findings related to PrP levels, PrPsc deposition, spongiform degeneration, neuron loss, astrogliosis and microgliosis in sCJD cases in the present series
Neuropathological characterization of sCJD MM1 and VV2 subtypes
Neuropathological examination showed variable severity of degeneration with a transcortical distribution in the frontal cortex of MM1 cases, whereas a preferential laminar distribution mainly involving the deep layers occurred in the frontal cortex of VV2 cases. Purkinje and granule cell layers of the cerebellum were largely preserved in MM1 but the cerebellum of VV2 cases was much more affected and the three cortical layers were severely involved. Spongiform degeneration composed of micro-vacuoles was observed in the frontal cortex of sCJD MM1 and VV2 subtypes and in the cerebellum of sCJD MM1. Medium-size vacuoles were also detected in the frontal cortex and cerebellum in sCJD VV2 (). Marked astrogliosis was observed on histological sections. In addition, GFAP expression of total homogenates was increased in the frontal cortex and cerebellum in sCJD cases when compared with controls ().
Figure 4. Subtype-specific neuropathological changes, astrogliosis and inflammation in sCJD brain samples. (A) Haematoxylin and eosin staining of sCJD MM1 and sCJD VV2 cases from frontal cortex and cerebellum showing spongiform degeneration. (B) Western blot analysis of GFAP levels in the frontal cortex and cerebellum in control, sCJD MM1 and sCJD VV2 in five representative cases reveals a marked increase in GFAP levels in disease states. Densiometric values of GFAP in all the cases analyzed by western blot: control (n = 15), sCJD MM1 (n = 15), sCJD VV2 (n = 14) illustrates the significant increase of GFAP in the frontal cortex and cerebellum in MM1 and VV2 samples. (C) CD-68 immunoreactivity reflects progressive stages of microglia activation (upper panels) and increased numbers of microglial cells in the frontal cortex and cerebellum in MM1 and VV2. (D) mRNA average expression levels of IL6 and TNF-α in the frontal cortex and cerebellum in control, sCJD MM1 and sCJD VV2 cases, as determined by specific TaqMan PCR assays. Significant increase of TNF-α values are observed in the frontal cortex and cerebellum in sCJD MM1 and sCJD VV2, whereas a significant increase in the expression of IL-6 mRNA was seen in the frontal cortex in sCJD MM1 and cerebellum in sCJD VV2. IL-6 and TNF-α mRNA are normalized using GUSβ as internal control (housekeeping). *P > 0.05; **P > 0.01; ***P > 0.001: control vs sCJD, #P > 0.05: sCJD MM1 vs VV2. AU: arbitrary units.
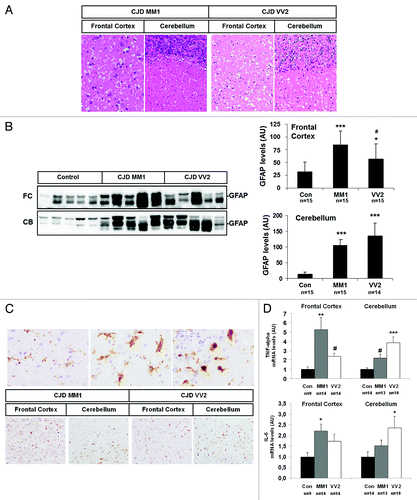
Variable microglial reaction, as revealed with the anti-CD68 antibody, occurred in the frontal cortex and cerebellum in MM1 and VV2 cases (). This was accompanied by increased mRNA expression of two inflammatory markers, IL-6 (a cytokine that stimulates immune response) and TNF-α (a cytokine involved in systemic inflammation), in sCJD. Interestingly, higher levels of both cytokines were detected in the frontal cortex in sCJD MM1 and in the cerebellum in sCJD VV2 (). A summary of the findings related to PrP levels, PrPsc deposition, spongiform degeneration, neuron loss, and gliosis is shown in .
PrP mRNA and PrPc in the frontal cortex in AD and LBD
In order to analyze whether low expression of PrP mRNA in sCJD was disease-specific, PrP mRNA was assessed by RT-qPCR in frontal cortex of AD (stages III–IV and V–VI), LBD (stages 3–6) and age-matched control samples. Decreased PrP mRNA was found in frontal cortex of AD (V–VI) and LBD (3–6) when compared with controls (). No significant differences were noticed between control and AD cases at earlier stages of the disease (stages III–IV). Similar findings were obtained using two different housekeeping genes for normalization. The same samples were analyzed by western blotting. Densitometric values did not show significant differences between control, AD (V–VI) and LBD cases (). As expected, PK digestion of AD (V–VI) and LBD samples confirmed that PrP in AD and LBD was not due to the presence of pathogenic forms of PrPsc ().
Figure 5. Expression of PrP in frontal cortex in AD and LBD. (A) mRNA average expression levels of PrP in the frontal cortex of control, AD (III-IV), AD (V-VI) and LBD cases, as determined by TaqMan PCR assays. Values for PrP mRNA are normalized using GUSβ and XPNPEP1 as internal controls. Reduced PrP mRNA levels are seen in AD (V-VI) and LBD. (B) western blot analysis of PrP in the frontal cortex of control and AD (III-IV), AD (V-VI) and LBD cases. Five representative cases are shown; β-actin is used to normalize total protein loaded onto the gel. Densiometric values of all the cases analyzed by western blot: control (n = 14), AD (III-IV) (n = 14), AD (V-VI) (n = 14) and LBD (n = 14) reveals no differences in PrP expression levels between control and diseased cases. *P > 0.05; **P > 0.01; ***P > 0.001: control vs AD/LBD cases. AU: arbitrary units. (C) PK digestion of AD and LBD samples confirms that AD and LBD do not contain PrPsc.
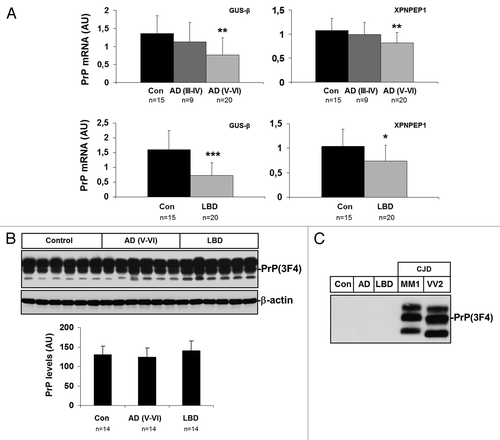
Immunohistochemistry analysis of AD and LBD cases with anti-PrP antibodies demonstrated accumulation of PrPc in neurites surrounding β-amyloid plaques in both diseases, as previously observed.Citation18
PrP mRNA and PrPc in the frontal cortex in FTLD (familial FTLD-tau and FTLD-TDP-43), and PSP
Cases with FTLD showed reduced mRNA PrP expression with both housekeeping genes. However, PrP mRNA levels in PSP were significantly reduced only when analyzed using GUSβ as housekeeping gene ().
Figure 6. Expression of PrP in PSP and FTLD (fFTLD-tau and FTLD-TDP-43) frontal cortex. (A) mRNA average expression levels of PrP in the frontal cortex in control (Con), PSP and FTLD cases determined by TaqMan PCR assays. Values for PrP mRNA are normalized using GUSβ and XPNPEP1 as internal controls. (B) western blot analysis of PrP in the frontal cortex of control (Con), PSP and FTLD cases in five representative cases per condition; β-actin is used to normalize total protein loading. Densiometric values of all the cases analyzed by western blot: control (n = 7), PSP (n = 12) and FTLD (n = 7) show reduced PrP expression in PSP and FTLD. *P > 0.05; **P > 0.01; ***P > 0.001: control vs PSP/FTLD cases. (C) PK digestion of PSP and FTLD samples confirm that PSP and FTLD do not contain PrPsc.
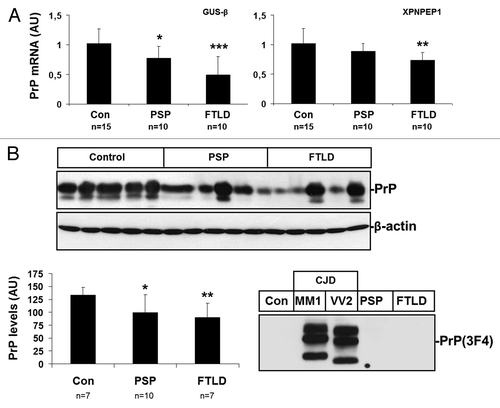
PrP protein levels, as revealed by western blotting, were reduced in frontal cortex in PSP and FTLD cases when compared with controls (). PK digestion of PSP and FTLD samples confirmed that PSP and FTLD did not contain PrPsc ().
Discussion
The present findings show decreased PrP mRNA expression in the frontal cortex in sCJD MM1 and cerebellum in sCJD VV2, and in the frontal cortex in LBD, PSP, FTLD and advanced stages of AD. Reduced PrP mRNA expression is accompanied by decreased values of total PrP protein in the frontal cortex in sCJD MM1 and by increased values of total PrP protein in the cerebellum in sCJD VV2. PrP protein levels in the frontal cortex in AD and LBD do not differ from those seen in age-matched controls, whereas PrP protein levels are reduced in the frontal cortex in PSP and FTLD. Therefore, no match exists between PrP mRNA and protein levels in several neurodegenerative diseases, as well as in different brain regions regarding, the later, sCJD subtypes MM1 and VV2. Although the common decrease in PrP mRNA in frontal cortex in several neurodegenerative diseases with brain involvement may be related to neuronal degeneration, the variability in PrP protein levels is likely modulated by variegated disease-dependent mechanisms. Similarly, a low correlation between PrP mRNA and PrPsc has been observed in sheep experimentally infected with scrapie, as low levels of PrP mRNA and high PrPsc accumulation are detected in the spleen.Citation19
Differences of PrPsc deposits in MM1 and VV2 remain obscure, and they are not directly related to either PrP mRNA or total PrP protein levels. PrPc is mainly mono-glycosylated and di-glycosylated in control brains while a significant increase of un-glycosylated forms is observed in sCJD. Glycosylation differences between normal and pathogenic prion protein isoforms are observed in Syrian hamsters,Citation20 and regulation of glycosylation has been proposed as a modulator in the formation of PrPsc.Citation21 In addition, PrP glycosylation determines the outcome of the disease in infected mice, as well as the strain-specific properties of the infection.Citation22,Citation23 An additional observation is the lack of relationship between PrPsc deposition and nerve cell damage and spongiform degeneration in sCJD excepting the cerebellum in sCJD VV2. Lack of correlation between abnormal PrP depositions and loss of synaptic function, and associated neurological deficits has also been reported in human and animal prion diseases.Citation24-Citation26 In contrast, PrP protein levels in sCJD correlate with astrogliosis and neuroinflammation represented by microglial and interleukine responses. It is feasible that PrP intermediates including non-fibrillar oligomers rather than PrPsc deposits are the main neurotoxic agents in sCJD, as has been demonstrated in primary cultures of neurons, and in vivo in PrPsc-injected mice.Citation27,Citation28
Regarding AD and LBD, preserved brain PrP protein levels in spite of decreased PrP mRNA may be explained by the reduced turnover of PrP protein mainly due to the accumulation of PrPc in dystrophic neurites surrounding β-amyloid plaques.Citation18 It can be speculated that PrPc deposition in β-amyloid plaques in AD and LBD can be explained by the interactions between PrPc and β-amyloid oligomers.Citation29,Citation30 Indeed, PrPc has been demonstrated to be a co-factor in the neurotoxic effect of oligomeric β-amyloid.Citation31 Finally, a direct connectivity on the signaling pathways between PrPc-Fyn activation, oligomeric β-amyloid and tau hyperphosphorylation has been reported.Citation32
The observation of reduced PrP mRNA expression levels in the brain in several neurodegenerative diseases including sCJD are in accordance with decreased PrPc protein levels in CSF.Citation16 Together, these findings suggest that low PrP protein levels in the CSF correlate with PrP mRNA expression rather than with total PrP protein levels in the brains of patients with neurodegenerative diseases of the CNS, including sCJD, and they probably correlate with the severity of brain damage.
Material and Methods
Cases and general processing
Brain tissue was obtained from the Institute of Neuropathology Brain Bank (HUB-ICO-IDIBELL Biobank) and the Biobank of Hospital Clinic-IDIBAPS following the guidelines of the Spanish legislation and the local ethics committees. The post-mortem interval between death and tissue processing was between 1 h 45 min and 24 h 30 min. For CJD cases, the whole brain excepting one sample of the frontal pole and one cerebellar hemisphere, was fixed in 4% buffered formalin and then treated with formic acid. The fresh samples of the frontal lobe and cerebellum were rapidly frozen and stored at −80 °C until use. For the other cases, including controls, one hemisphere was immediately cut in coronal sections, 1 cm thick, and selected areas of the encephalon were rapidly dissected, frozen on metal plates over dry ice, placed in individual air-tight plastic bags, numbered with water-resistant ink, and stored at −80 °C until use for biochemical studies. The other hemisphere was fixed by immersion in 4% buffered formalin for 3 weeks for morphological studies. Neuropathological examination was performed in every case on paraffin-embedded samples and cut with a sliding microtome. Tissue sections stained with hematoxylin and eosin, Klüver-Barrera, or processed for inmunohistochemistry to detect the presence of phospho-tau (AT8 from Innogenetics), β-amyloid (βA4-amyloid, Roche), α-synuclein (AB5038 from Chemicon), ubiquitin (Z458 from Dako), TDP-43 (2E2-D3 from Abnova), glial fibrillary acidic protein (GFAP, Dako), CD68 (Dako), PrP (clone 3F4, Millipore) and αB-crystallin (NCL-ABCrys-512 from Novocastra). A summary of the cases and their use in the different studies is shown in .
Table 2. Cases studied in the present series
Semi-quantitative assessment of spongiform change, neuronal loss, astrogliosis and microglial proliferation in sCJD was performed on frontal cortex and cerebellum. Parameters were scored as 0 = absent, 1 = mild, 2 = moderate and 3 = severe. Mild, moderate and severe were considered when magnifications of × 200, × 100, and × 20 were needed to see the lesions.
CSF samples were obtained from an unrelated series of patients with sCJD and controls. All individuals in this group were deceased and postmortem neuropathological examination confirmed the diagnosis: sCJD MM1 n = 33, sCJD VV2 n = 8. Control cases (n = 34) had lumbar punctures with normal CSF findings. Clinical symptoms that indicate the practice of a lumbar puncture for diagnostic purposes in the control group were headache, vertigo, and polyneuropathy of unknown origin.
All the biochemical studies were performed in P3 biosafety rooms.
CJD subtype characterization
The analysis of the codon 129 genotype of PrP gene (Met or Val) was performed after isolation of genomic DNA from blood samples according to standard methods.Citation33 western blot profile of PrPsc was classified as type 1 (unglycosylated PrPsc of 20/21 kDa) or type 2 (unglycosylated PrPsc of 19 kDa) based on electrophoretic mobility after proteinase K (PK) digestion.Citation5,Citation7
RNA purification
The purification of RNA of frontal cortex of AD, LBD, FTLD, PSP cases and age-matched controls was performed with RNeasy Lipid Tissue Mini Kit (Qiagen 74804) following the protocol provided by the supplier. During purification, samples were treated with RNase-free DNase set (Qiagen 79254) for 15 min to avoid extraction and later amplification of genomic DNA. The purification of RNA from cerebellum and frontal cortex of CJD and age-matched controls was performed using miRvana isolation kit (Ambion AM1560) according to the manufacturer’s instructions. After purification, samples were treated with DNase-free kit (Ambion AM1906) for 30 min to avoid the extraction and the subsequent amplification of genomic DNA. The concentration of each sample was determined at 340 nm using NanoDrop 2000 spectrophotometer (Thermo Scientific). RNA integrity number (RIN) was verified with the Agilent 2100 Bioanalyzer (Agilent), and the threshold for sample selection was set at RIN equal or higher than 6.
Retrotranscription reaction
The retrotranscriptase reaction of the RNA samples was performed with the High Capacity cDNA Archive kit (Applied Biosystems) following the protocol provided by the manufacturer and using the Gene Amp® 9700 PCR System thermocycler (Applied Biosystems). A parallel reaction for a RNA sample was run in the absence of reverse transcriptase to assess the degree of contaminating genomic DNA.
RT-qPCR
PCR assays were conducted in duplicate on cDNA samples obtained from the retrotranscription reaction and diluted 1:30 for AD, LBD, FTLD, and PSP samples and their controls, and diluted 1:20 for CJD samples and their controls in 384-well optical plates (Applied Biosystems) utilizing an ABI Prism 7900 Sequence Detection System (Applied Biosystems). Parallel amplification reactions for each sample were performed using the 20 × TaqMan Gene Expression Assays (Applied Biosystems) and 2 × TaqMan Universal PCR Master Mix (Applied Biosystems). β-glucuronidase (GUSβ) and X-prolyl aminopeptidase P1 (XPNPEP1) were used as internal housekeeping gene controls for normalization. The reactions were performed with the following parameters: 50 °C for 2 min, 95 °C for 10 min, and 40 cycles of 95 °C for 15 s and 60 °C for 1 min. TaqMan PCR data were captured using the Sequence Detector Software (SDS version 2.1, Applied Biosystems).
Subsequently, CT data for each sample were analyzed with the double delta CT (ΔΔCT) method. Delta CT (ΔCT) values represent the normalized levels of each target gene in relation to endogenous controls (GUSβ and XPNPEP1), whereas ΔΔCT values were calculated as the ΔCT of each sample minus the mean ΔCT of the population of control samples (calibrator samples). The fold change was determined using the equation 2−ΔΔCT. Mean fold change values of each group were compared with one-way ANOVA followed by Tukey’s test using the Statgraphics Statistical Analysis and Data Visualization Software version 5.1. Differences between groups were considered statistically significant at *P < 0.05, ** P < 0.01, *** P < 0.001. Probes used in this study: GUS-β: GCTACTACTT GAAGATGGTG ATCGC, XPNPEP1: CAAAGAGTGC GACTGGCTCA ACAAT, PrP: CGACCGAGAG CAGTCATTAT GGCGA.
Western blotting
Human tissues were lysed in Lysis Buffer: 100 mM Tris pH 7, 100 mM NaCl, 10 mM EDTA, 0.5% NP-40 and 0.5% Sodium Deoxycolate plus protease and phosphatase inhibitors. After centrifugation at 14 000 g for 20 min at 4 °C, supernatants were quantified for protein concentration (BCA, Pierce), mixed with SDS-PAGE sample buffer, boiled, and subjected to 8–15% SDS-PAGE. Gels were transferred onto nitrocellulose membranes, processed for specific immunodetection using one of the following primary antibodies: 3F4 antibody (Millipore: MAB1562), anti-GFAP (Dako: N1506) and anti-β-actin (Sigma: A2228). The immunoreaction was visualized by chemiluminescence (ECL Amersham) Densitometries were performed with ImageJ software and values were normalized using β-actin (non-CJD samples), and β-actin and Commassie Blue staining (CJD samples). Some samples were pre-incubated with proteinase K (Sigma: P2308) or incubated with PNGase F (New England Biolabs P0704).
ELISA assays
CSF samples were obtained by lumbar puncture. Routine investigation of the CSF did not reveal significant abnormalities with respect to the cell counts and proteins. Blood-stained CSF samples were excluded from analysis because contamination of this type can lead to “false positive” results. We used Platelia BSE-Detection Kit (Bio-Rad Laboratories GmbH) according to the instructions of the supplier. This kit is based on ELISA techniques and is employed as a rapid BSE test for qualitative determination of PrPsc in the brain tissue of cattle and sheep. For our experiments, we omitted the PK digestion step, which digests PrPc but not PrPsc, since our aim was to determine total PrP levels in the CSF (in subjects with prion diseases, PrPc plus potentially extremely low amount of PrPsc). CSF samples were applied undiluted. Calibration was performed with recombinant PrP (Prionics) and a calibration curve was calculated. This curve was used to determine the PrP concentrations based on the values obtained from the ELISA assay. The signal was measured at 450 nm with 1420 Multilabel Counter Victor 2 (Wallac).
| Abbreviations: | ||
| sCJD | = | sporadic Creutzfeldt-Jakob disease |
| AD | = | Alzheimer disease |
| LBD | = | Lewy body disease |
| PSP | = | progressive supranuclear palsy |
| FTLD | = | frontotemporal lobar degeneration |
| CSF | = | cerebrospinal fluid |
| CNS | = | central nervous system |
| PK | = | proteinase K |
Additional material
Download Zip (560.5 KB)Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
This study was funded by the Seventh Framework Program of the European Commission DEVELAGE project, the European Union Joint Program DEMTEST, the Spanish Ministry of Health, Instituto Carlos III: FIS PI1100968, and CIBERNED project BESAD-P. We are grateful for the support for previous studies in the context of the BrainNet Europe II, LSHM-CT-2004-503039. Supply of brain samples from the Biobank of Hospital Clinic-IDIBAPS is acknowledged. We thank all brain donors and relatives for generous brain donation for research, as well as referring physicians; Dr Judith Navarro and Carina Antiga for their support in the Brain Donor Program; Rosa Ribera, Sara Charif, Abel Muñoz, Leire Etxarri, Esteban González and Jesús González for their technical support; and T Yohannan for editorial assistance. E Gelpí is partly funded by the Spanish “Ministerio de Economía y Competitividad, Subprograma Técnicos de Apoyo, 2011.”
References
- Aguzzi A, Sigurdson C, Heikenwaelder M. Molecular mechanisms of prion pathogenesis. Annu Rev Pathol 2008; 3:11 - 40; http://dx.doi.org/10.1146/annurev.pathmechdis.3.121806.154326; PMID: 18233951
- Colby DW, Prusiner SB. Prions. Cold Spring Harb Perspect Biol 2011; 3:a006833; http://dx.doi.org/10.1101/cshperspect.a006833; PMID: 21421910
- Aguzzi A, Heikenwalder M, Polymenidou M. Insights into prion strains and neurotoxicity. Nat Rev Mol Cell Biol 2007; 8:552 - 61; http://dx.doi.org/10.1038/nrm2204; PMID: 17585315
- Parchi P, Castellani R, Capellari S, Ghetti B, Young K, Chen SG, Farlow M, Dickson DW, Sima AA, Trojanowski JQ, et al. Molecular basis of phenotypic variability in sporadic Creutzfeldt-Jakob disease. Ann Neurol 1996; 39:767 - 78; http://dx.doi.org/10.1002/ana.410390613; PMID: 8651649
- Parchi P, Giese A, Capellari S, Brown P, Schulz-Schaeffer W, Windl O, Zerr I, Budka H, Kopp N, Piccardo P, et al. Classification of sporadic Creutzfeldt-Jakob disease based on molecular and phenotypic analysis of 300 subjects. Ann Neurol 1999; 46:224 - 33; http://dx.doi.org/10.1002/1531-8249(199908)46:2<224::AID-ANA12>3.0.CO;2-W; PMID: 10443888
- Head MW, Ironside JW. Review: Creutzfeldt-Jakob disease: prion protein type, disease phenotype and agent strain. Neuropathol Appl Neurobiol 2012; 38:296 - 310; http://dx.doi.org/10.1111/j.1365-2990.2012.01265.x; PMID: 22394291
- Parchi P, Saverioni D. Molecular pathology, classification, and diagnosis of sporadic human prion disease variants. Folia Neuropathol 2012; 50:20 - 45; PMID: 22505361
- Puoti G, Bizzi A, Forloni G, Safar JG, Tagliavini F, Gambetti P. Sporadic human prion diseases: molecular insights and diagnosis. Lancet Neurol 2012; 11:618 - 28; http://dx.doi.org/10.1016/S1474-4422(12)70063-7; PMID: 22710755
- Herms J, Tings T, Gall S, Madlung A, Giese A, Siebert H, Schürmann P, Windl O, Brose N, Kretzschmar H. Evidence of presynaptic location and function of the prion protein. J Neurosci 1999; 19:8866 - 75; PMID: 10516306
- Büeler H, Aguzzi A, Sailer A, Greiner RA, Autenried P, Aguet M, Weissmann C. Mice devoid of PrP are resistant to scrapie. Cell 1993; 73:1339 - 47; http://dx.doi.org/10.1016/0092-8674(93)90360-3; PMID: 8100741
- Riemenschneider M, Wagenpfeil S, Vanderstichele H, Otto M, Wiltfang J, Kretzschmar H, Vanmechelen E, Förstl H, Kurz A. Phospho-tau/total tau ratio in cerebrospinal fluid discriminates Creutzfeldt-Jakob disease from other dementias. Mol Psychiatry 2003; 8:343 - 7; http://dx.doi.org/10.1038/sj.mp.4001220; PMID: 12660807
- Sanchez-Juan P, Green A, Ladogana A, Cuadrado-Corrales N, Sáanchez-Valle R, Mitrováa E, Stoeck K, Sklaviadis T, Kulczycki J, Hess K, et al. CSF tests in the differential diagnosis of Creutzfeldt-Jakob disease. Neurology 2006; 67:637 - 43; http://dx.doi.org/10.1212/01.wnl.0000230159.67128.00; PMID: 16924018
- Weber T, Otto M, Bodemer M, Zerr I. Diagnosis of Creutzfeldt-Jakob disease and related human spongiform encephalopathies. Biomed Pharmacother 1997; 51:381 - 7; http://dx.doi.org/10.1016/S0753-3322(97)89430-9; PMID: 9452787
- Zerr I, Bodemer M, Gefeller O, Otto M, Poser S, Wiltfang J, Windl O, Kretzschmar HA, Weber T. Detection of 14-3-3 protein in the cerebrospinal fluid supports the diagnosis of Creutzfeldt-Jakob disease. Ann Neurol 1998; 43:32 - 40; http://dx.doi.org/10.1002/ana.410430109; PMID: 9450766
- Wong BS, Green AJ, Li R, Xie Z, Pan T, Liu T, Chen SG, Gambetti P, Sy MS. Absence of protease-resistant prion protein in the cerebrospinal fluid of Creutzfeldt-Jakob disease. J Pathol 2001; 194:9 - 14; http://dx.doi.org/10.1002/path.872; PMID: 11329135
- Meyne F, Gloeckner SF, Ciesielczyk B, Heinemann U, Krasnianski A, Meissner B, Zerr I. Total prion protein levels in the cerebrospinal fluid are reduced in patients with various neurological disorders. J Alzheimers Dis 2009; 17:863 - 73; PMID: 19542614
- Torres M, Cartier L, Matamala JM, Hernández N, Woehlbier U, Hetz C. Altered Prion protein expression pattern in CSF as a biomarker for Creutzfeldt-Jakob disease. PLoS One 2012; 7:e36159; http://dx.doi.org/10.1371/journal.pone.0036159; PMID: 22558368
- Esiri MM, Carter J, Ironside JW. Prion protein immunoreactivity in brain samples from an unselected autopsy population: findings in 200 consecutive cases. Neuropathol Appl Neurobiol 2000; 26:273 - 84; http://dx.doi.org/10.1046/j.1365-2990.2000.00239.x; PMID: 10886685
- Sørby R, Austbø L, Press CM, Skretting G, Landsverk T, Espenes A. PrP expression, PrPSc accumulation and innervation of splenic compartments in sheep experimentally infected with scrapie. PLoS One 2009; 4:e6885; http://dx.doi.org/10.1371/journal.pone.0006885; PMID: 19727393
- Rudd PM, Endo T, Colominas C, Groth D, Wheeler SF, Harvey DJ, Wormald MR, Serban H, Prusiner SB, Kobata A, et al. Glycosylation differences between the normal and pathogenic prion protein isoforms. Proc Natl Acad Sci U S A 1999; 96:13044 - 9; http://dx.doi.org/10.1073/pnas.96.23.13044; PMID: 10557270
- Winklhofer KF, Heller U, Reintjes A, Tatzelt J. Inhibition of complex glycosylation increases the formation of PrPsc. Traffic 2003; 4:313 - 22; http://dx.doi.org/10.1034/j.1600-0854.2003.00088.x; PMID: 12713659
- Cancellotti E, Bradford BM, Tuzi NL, Hickey RD, Brown D, Brown KL, Barron RM, Kisielewski D, Piccardo P, Manson JC. Glycosylation of PrPC determines timing of neuroinvasion and targeting in the brain following transmissible spongiform encephalopathy infection by a peripheral route. J Virol 2010; 84:3464 - 75; http://dx.doi.org/10.1128/JVI.02374-09; PMID: 20106922
- Cancellotti E, Mahal SP, Somerville R, Diack A, Brown D, Piccardo P, Weissmann C, Manson JC. Post-translational changes to PrP alter transmissible spongiform encephalopathy strain properties. EMBO J 2013; 32:756 - 69; http://dx.doi.org/10.1038/emboj.2013.6; PMID: 23395905
- Ferrer I, Puig B, Blanco R, Martí E. Prion protein deposition and abnormal synaptic protein expression in the cerebellum in Creutzfeldt-Jakob disease. Neuroscience 2000; 97:715 - 26; http://dx.doi.org/10.1016/S0306-4522(00)00045-2; PMID: 10842016
- Ferrer I, Rivera R, Blanco R, Martí E. Expression of proteins linked to exocytosis and neurotransmission in patients with Creutzfeldt-Jakob disease. Neurobiol Dis 1999; 6:92 - 100; http://dx.doi.org/10.1006/nbdi.1998.0226; PMID: 10343324
- Jeffrey M, McGovern G, Sisó S, González L. Cellular and sub-cellular pathology of animal prion diseases: relationship between morphological changes, accumulation of abnormal prion protein and clinical disease. Acta Neuropathol 2011; 121:113 - 34; http://dx.doi.org/10.1007/s00401-010-0700-3; PMID: 20532540
- Corsaro A, Thellung S, Villa V, Nizzari M, Florio T. Role of prion protein aggregation in neurotoxicity. Int J Mol Sci 2012; 13:8648 - 69; http://dx.doi.org/10.3390/ijms13078648; PMID: 22942726
- Simoneau S, Rezaei H, Salès N, Kaiser-Schulz G, Lefebvre-Roque M, Vidal C, Fournier JG, Comte J, Wopfner F, Grosclaude J, et al. In vitro and in vivo neurotoxicity of prion protein oligomers. PLoS Pathog 2007; 3:e125; http://dx.doi.org/10.1371/journal.ppat.0030125; PMID: 17784787
- Laurén J, Gimbel DA, Nygaard HB, Gilbert JW, Strittmatter SM. Cellular prion protein mediates impairment of synaptic plasticity by amyloid-beta oligomers. Nature 2009; 457:1128 - 32; http://dx.doi.org/10.1038/nature07761; PMID: 19242475
- Um JW, Nygaard HB, Heiss JK, Kostylev MA, Stagi M, Vortmeyer A, Wisniewski T, Gunther EC, Strittmatter SM. Alzheimer amyloid-β oligomer bound to postsynaptic prion protein activates Fyn to impair neurons. Nat Neurosci 2012; 15:1227 - 35; http://dx.doi.org/10.1038/nn.3178; PMID: 22820466
- Kudo W, Lee HP, Zou WQ, Wang X, Perry G, Zhu X, Smith MA, Petersen RB, Lee HG. Cellular prion protein is essential for oligomeric amyloid-β-induced neuronal cell death. Hum Mol Genet 2012; 21:1138 - 44; http://dx.doi.org/10.1093/hmg/ddr542; PMID: 22100763
- Larson M, Sherman MA, Amar F, Nuvolone M, Schneider JA, Bennett DA, Aguzzi A, Lesné SE. The complex PrP(c)-Fyn couples human oligomeric Aβ with pathological tau changes in Alzheimer’s disease. J Neurosci 2012; 32:16857 - 71a; http://dx.doi.org/10.1523/JNEUROSCI.1858-12.2012; PMID: 23175838
- Windl O, Giese A, Schulz-Schaeffer W, Zerr I, Skworc K, Arendt S, Oberdieck C, Bodemer M, Poser S, Kretzschmar HA. Molecular genetics of human prion diseases in Germany. Hum Genet 1999; 105:244 - 52; http://dx.doi.org/10.1007/s004390051096; PMID: 10987652