Abstract
Prion diseases comprise a group of fatal neurodegenerative disorders characterized by the autocatalytic conversion of the cellular prion protein PrPC into the infectious misfolded isoform PrPSc. Increasing evidence supports a specific role of oxidative stress in the onset of pathogenesis. Although the associated molecular mechanisms remain to be elucidated in detail, several studies currently suggest that methionine oxidation already detected in misfolded PrPSc destabilizes the native PrP fold as an early event in the conversion pathway. To obtain more insights about the specific impact of surface-exposed methionine residues on the oxidative-induced conversion of human PrP we designed, produced, and comparatively investigated two new pseudosulfoxidation mutants of human PrP 121–231 that comprises the well-folded C-terminal domain. Applying circular dichroism spectroscopy and dynamic light scattering techniques we showed that pseudosulfoxidation of all surface exposed Met residues formed a monomeric molten globule-like species with striking similarities to misfolding intermediates recently reported by other groups. However, individual pseudosulfoxidation at the polymorphic M129 site did not significantly contribute to the structural destabilization. Further metal-induced oxidation of the partly unfolded pseudosulfoxidation mutant resulted in the formation of an oligomeric state that shares a comparable size and stability with PrP oligomers detected after the application of different other triggers for structural conversion, indicating a generic misfolding pathway of PrP. The obtained results highlight the specific importance of methionine oxidation at surface exposed residues for PrP misfolding, strongly supporting the hypothesis that increased oxidative stress could be one causative event for sporadic prion diseases and other neurodegenerative disorders.
Introduction
Prion diseases comprise a group of fatal neurodegenerative disorders that affect humans and animals. The associated histopathological features, such as spongiform brain degeneration, represent the main consequences of the cerebral deposition of a misfolded isoform (PrPSc) of the cellular prion protein (PrPC), often into amyloid plaques.Citation1 Human PrPC is a soluble and monomeric cell surface sialoglycoprotein that exhibits significant susceptibility toward proteinase K (PK) digestion. The globular C-terminal domain adopts a predominantly α-helical fold, whereas the N-terminal part is considered to be largely unstructured.Citation2 Conversely, PrPSc resembles an insoluble aggregated protein multimer characterized by an enhanced PK resistance and an increased amount of β-sheet conformation.Citation3 According to the widely accepted “protein-only” hypothesis, PrPSc is the infectious and transmissible agent, which serves as a template for the transition of PrPC on the pathway of a self-propagating autocatalytic conversion process.Citation4 About 85% of all known human prion cases arise spontaneously, whereas mutations within the folded C-terminal domain of PrPC provoke an inherited form of Creutzfeld-Jakob disease.Citation5 An acquired variant (vCJD) has been linked to the transmission of the infectious agent by contaminated beef.Citation6
Increasing evidence suggests a causal link between oxidative stress and prion diseases, supporting a protective role of PrPC by acting as an antioxidant.Citation7,Citation8 In proteins, particularly the sulfur containing side chains of cysteine (Cys) and methionine (Met) residues are most sensitive to oxidation by highly reactive oxygen species (ROS).Citation9 Copper redox cycling in the presence of physiological reductants, e.g., ascorbate, is the main source of ROS formation in the organisms, converting the moderate hydrophobic thioether side chain of methionines into a more hydrophilic sulfoxide that frequently affects the structural integrity of proteins.Citation9 In healthy tissue this chemical posttranslational modification is usually reversed by cellular methionine sulfoxide reductases.Citation10 However, in older ages, the loss of protection mechanisms due to a compromised activity of recycling enzymes induces an imbalance that results in increased methionine oxidation, which is observed in an accumulating number of proteins.Citation9,Citation11 Methionine sulfoxidation is known to be involved in positive cell signaling, but may also lead to the onset of protein misfolding diseases.Citation11 Moreover, an endogenous scavenger role was attributed to methionines, protecting vital sites of other proteins from damage as a first defense mechanism.Citation12
Mammalian PrPC is particularly prone to oxidation due to its unique high number of Met residues. Indeed, Met-sulfoxides have frequently been detected in brain deposited PrPSc isoforms and are currently discussed to be a distinct and specific signature for pathogenic prion proteins.Citation13-Citation15 Since minor stochastically formed covalent modifications of the PrP polypeptide chain by environmental factors are assumed to be the initial event within the misfolding cascade,Citation16 Met oxidation is now considered to be one destabilizing trigger that causes the cascade of events leading to PrP misfolding and formation of toxic species in prion diseases.Citation16-Citation19 In the structured globular domain of human PrP (residues 125–231), Met129, Met134, Met154, and Met166 are surface exposed and therefore accessible to immediate oxidation, whereas Met205 and Met206 are completely and Met213 is partially buried. Several recent studies highlight that in particular the oxidation of the (partially) buried methionines located within helix 3 (Met205, Met206, and Met213) perturbs the hydrophobic core of PrP, resulting in the formation of monomeric molton globule-like species with a high aggregation propensity that could represent folding intermediates on the conversion pathway.Citation15,Citation17-Citation20 However, slight conformational rearrangements are assumed to initially occur to provide accessibility of the so far buried helix 3 methionines to ROS. The consequences of further structural destabilization depend on the experimental conditions, including the formation of amorphous aggregates, of β-sheet enriched oligomers, or of amyloid fibrils, as well as β-cleavage.Citation16,Citation17,Citation19,Citation21-Citation24 Another prominent relevance for destabilizing the native PrP fold was attributed by mutagenesis studies to polymorphic Met129 (M129V),Citation25 which modulates the risk and phenotype of prion diseases in humans.Citation26
We recently reported that both UV light- and metal-induced oxidation significantly affect the structural integrity of recombinant PrP.Citation21,Citation27 Depending on the number of oxidation susceptible residues that varies between the sequences of prion proteins from different mammalian species, two independent aggregation pathways have been observed, resulting in complete protein unfolding as well as in prion-like structural conversion into soluble β-oligomers, which share a high degree of identity with oligomeric species formed after application of other triggers.Citation28 In line with the previous studies of ours and others summarized above, the aim of the present study is to systematically extend the investigation of the impact of methionine oxidation on the conversion process of human PrP. Since the high number of Met residues within human PrP prevents to confer site-specificity in the changes using chemical oxidation approaches, we focused this study on the comparison of metal-induced oxidation of wt human PrP (121–231) with that of pseudosulfoxidated PrP molecules in the context of previous mutagenesis experiments.Citation17,Citation18 By site-directed serine replacement of all surface exposed Met residues, we suggested to form an intermediate species that resembles the partially oxidation state immediately before the so far buried residues Met205 and Met206 become accessible for ROS. Additionally, we mimicked the sulfoxidation-based hydrophobicity increase at the polymorphic residue Met129 by site-directed threonine replacement. Thus, the previous PrP pseudosulfoxidation approaches were combined and extended in this study, forming folding intermediates that provide new opportunities for the investigation of the misfolding cascade of PrP.
Results
Design of human prion protein mutants
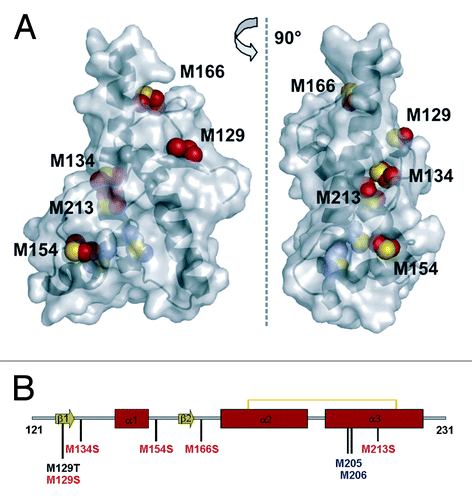
To investigate the impact of methionine oxidation on the oxidative-induced conversion process of PrP, we focused on wild type human PrP 121–231, which only comprises the well-folded C-terminal domain that is directly involved in the conformational change. Previous studies reported that the unstructured N-terminal domain did not significantly influence the destabilization of the native PrP fold if induced by site-specific mutation or by oxidation.Citation19,Citation25 Additionally, we designed two pseudosulfoxidation mutants to mimic methionine oxidation at selected, surface exposed Met residues, which should enable a site-specific investigation of the structural consequences of oxidation at solvent accessible methionines only. The strategy to provide a property of a chemical covalent modification by mutation against a suitable residue is well established to mimic phosphorylation and also sulfoxidation.Citation17,Citation29 Moreover, serine was already reported as a suitable mimic of MetO, since its hydroxyl group provides the polar charge of a sulfoxidized methionine while retaining its secondary structure propensities, even if the serine side chain is shorter than that of MetO.Citation17,Citation18 Based on wt hPrP 121–231, we thus generated a pseudosulfoxidation mutant containing the substitutions M129S-M134S-M154S-M166S-M213S (v-hPrP121–231), in which all surface exposed Met residues were replaced by serine residues (). The highly buried Met205 and Met206 residues that are already known to be essential for proper PrP folding were not included.Citation17 The use of Ser maintains the direct comparability of our results with that previously reported by Lisa et al. (2010),Citation17 who investigated the structural consequences of single M-to-S replacements (M134S, M154S, M206S, M213S) as well as of the double mutant M206S/M213S in hamster PrP. To evaluate the individual contribution of methionine oxidation at the polymorphic site Met129 to the structural destabilization of PrP, the site-specific mutant huPrP 121–231 M129T was additionally generated. Similar to Ser, threonine increases the local polarity, but mimics the structural demands of MetO better than Ser. Since the replacement with isosteric valine did not induce any structural instability at this position,Citation30 all resulting effects should be attributed only to the polarity increase within the M129T mutant.
Figure 1. Design of hPrP121–231 mutants. (A) NMR structure of the C-terminal domain of human PrP (residues 125–228, PDB 1QM0) in surface representation. Side chain atoms of solvent accessible methionine residues are highlighted as red spheres, while blue spheres indicate the completely buried Met residues 205 and 206. (B) Scheme of the secondary structure indicating the position of methionines. Met residues highlighted in blue are buried and have therefore not been considered for mutation, while the mutation shown in black resulted in the M129T mutant and the substitutions depicted in red were introduced to obtain the mutant v-hPrP121–231.
Expression and characterization of human prion protein mutants
The proteins were expressed as inclusion bodies in E. coli, purified, and oxidatively folded into the cellular α-isoform.Citation21 SDS-PAGE analysis showed pure protein bands at the expected molecular weight of approx. 13 kDa, confirming the chain integrity (). At a slightly acidic pH of 5.0, all solutions exhibited monodisperse behavior characterized by hydrodynamic protein radii (RH) of about 1.8 ± 0.1 nm (). Assuming a roughly globular shape, the observed RH values corresponded to theoretical values calculated from the molecular masses, indicating a monomeric state of all proteins.
Figure 2. Characterization of hPrP121–231 and its mutants. (A) SDS-PAGE analysis (Coomassie stain) of wt hPrP121–231, M129T mutant, and v-hPrP121–231 in the absence of reducing agents. (B) The conformational features of wt hPrP121–231 (black), hPrP121–231 M129T (blue) and v-hPrP121–231 (red) were probed by far-UV CD spectroscopy.
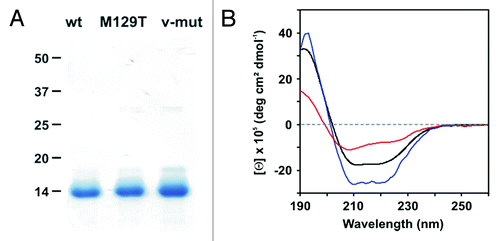
Table 1. Particle radii of wt hPrP 121–231 and its mutants determined by dynamic light scattering (DLS)
The conformational integrity of wt hPrP121–231 and its mutants was analyzed by far-UV CD spectroscopy. Distinct minima at a wavelength of 209 nm and 221 nm indicated a predominant α-helical fold of the wt protein and the M129T mutant, representing the typical conformation of cellular PrP ().Citation2 The M129T mutation seems to slightly stabilize the native state, as suggested by increased dichroic intensities compared with the wt spectrum, while the Θ205/Θ220 ratio remains similar. In contrast, the enhanced polarity at the M-to-S replacement sites of v-hPrP121–231 resulted in an overall flattened CD curve that exhibited a single predominant minimum shifted to lower wavelength. The associated increase in the Θ205/Θ220 ratio from 0.6 (wt and M129T mutant) to 1.2 strongly indicated that a partially unstructured state is formed. However, no indications for a gain in β-sheet structure as a consequence of reduced α-helix content have been obtained.
Structural conversion of wt hPrP121–231 by metal-induced oxidation
Recently we demonstrated that the site-specific oxidation of hPrP90–231, which resembles the PK resistant and amyloid forming core of PrPSc,Citation31 induced fundamental conformational changes including the formation of soluble, highly β-sheeted, and oligomeric PrP isoforms.Citation21 These results encouraged us to comparatively investigate the aggregation tendency of hPrP121–231 under oxidative conditions, directly illustrating the impact of the unstructured N-terminal part (residues 90 to 120). Aggregation kinetics were monitored using our established in vitro aggregation assays that mimics increasing cellular oxidative stress in solution by metal-induced protein oxidation.Citation21,Citation32
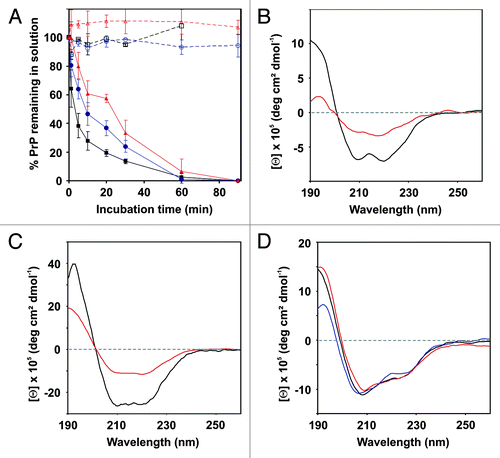
The exponential decrease of the protein concentration in the supernatants of wt hPrP121–231 samples exposed to oxidation revealed a rapid time-dependent aggregation at 37 °C, accompanied by heavy protein precipitation (). The corresponding half-life of 3.7 min was significantly decreased compared with the aggregation rate of hPrP90–231 (T½ = 30 min) determined under identical conditions.Citation21 After 60 min of oxidation, soluble wt hPrP121–231 was no longer detected in the supernatants. Since the PrP structure remained unaffected by the applied buffer and temperature conditions in the absence of metal-induced oxidation (; ), the observed aggregation process was directly linked to the proceeding copper-mediated redox reaction.
Figure 3. Metal-induced oxidation of hPrP121–231 and its mutants. (A) Time-resolved monitoring of wt-hPrP121–231 (black), hPrP121–231 M129T (blue), and v-hPrP121–231 (red) aggregation. Protein aliquots were incubated in the presence (straight line) and in the absence (dashed lines) of copper at 37°C for indicated time periods. Error bars represent standard deviations. (B–D) Far-UV CD spectroscopy monitoring changes in the secondary structure content of wt hPrP121–231 (B), hPrP121–231 M129T (C), and v-hPrP121–231 (D) caused by metal-induced oxidation. Spectra were recorded immediately at the beginning (black) as well as after 1 min (red) and 30 min (blue, only for v-hPrP121–231) of incubation under oxidative conditions.
In contrast to hPrP90–231,Citation21 distinct soluble and stable oligomeric isoforms have not been detected by DLS on the pathway of oxidative hPrP121–231 aggregation. Within the first minute of incubation the monomeric and homogeneous hPrP121–231 starts to assemble into large heterogeneous aggregates of high molecular weight (). SDS-PAGE analysis revealed that these aggregates are mainly stabilized by non-covalent interactions (data not shown). Small amounts of disulphide bridged dimers and trimers have only been detected in the later course of the oxidation reaction. Since EDTA treatment did not affect the aggregates, a contribution of protein bound copper ions to the stabilizing interactions was not indicated.
However, the aggregation process of wt hPrP121–231 was associated with profound secondary structure changes. The α-helical CD signal characteristic for cellular PrP changed within the first minute of oxidation into a flattened curve (). The associated decrease of the Θ205/Θ220 ratio from 0.6 to 0.4 together with the appearance of a single minimum around 215 nm indicated an increase in β-sheet structure. After 5 min of incubation, the soluble PrP concentration fell below the detection limit of the CD spectrometer due to the rapid aggregation process, preventing the determination of the secondary structure content at further time-points.
Structural conversion of hPrP121–231 mutants by metal-induced oxidation
Under identical oxidative conditions, the aggregation tendency of the M129T mutant (T½ = 8 min) and of v-hPrP121–231 (T½ = 24 min) is significantly reduced compared with that of wt hPrP121–231 (). Both proteins were not affected by the assay conditions in the absence of oxidative stress, correlating with a conserved overall structural stability. The increase in β-sheet structure also indicated for the M129T mutant after 1 min of oxidation was less pronounced compared with the wt protein, as shown by a minor decrease of the Θ205/Θ220 ratio from 0.60 to 0.55 (). Moreover, the partially unstructured, but still α-helix dominated fold of v-hPrP121–231 was detected by CD spectroscopy up to 30 min incubation under oxidative conditions without any obvious changes (), although only approximately 40% of v-hPrP121–231 remained in solution (). Further CD monitoring was prevented by the reduced PrP concentration in solution, as already observed for the M129T mutant after 5 min of incubation.
The exponential loss of soluble protein species indicates that oligomers and/or aggregates have also been formed during incubation of the M129T mutant and of v-hPrP121–231. However, the site-specific replacement of Met residues within hPrP121–231 caused alterations on the pathway of oxidative-induced aggregation compared with the wt protein. For the M129T mutant the degree of insoluble protein precipitation is reduced, but heterogeneous, non-covalently stabilized, high-MW aggregates were still detected, comparable with that of wt hPrP121–231 (). Removal of these aggregates by centrifugation resulted in the recovery of the corresponding monomeric DLS signal. Although the concentration of soluble protein is also exponentially decreased during oxidative incubation, the solution of v-hPrP121–231 showed no turbidity due to protein precipitation over the entire incubation period. Instead, an immediately formed stable and soluble PrP oligomer characterized by a hydrodynamic radius of approximately 16 nm was detected by a weak DLS signal, persisting up to 30 min in solution (). The determination of the secondary structure content of this oligomeric species by CD spectroscopy failed due to its low concentration in solution, which is in contrast to a 60% loss of soluble protein after 30 min of incubation. However, the sticky nature of the oligomeric species that we observed during handling and isolation trials could explain the unexpected protein loss.
Discussion
The elucidation of the molecular mechanisms that determine the conversion of PrP as well as the identification and characterization of the toxic and/or infectious species remain to be the most critical challenges within the field of prion research. As observed for other neurodegenerative disorders, prion diseases are frequently associated with the deposition of amyloid-like protein inclusions, which were first thought to mediate the misfolding-linked toxicity.Citation33 However, in the past years compelling evidence shifted the general focus in the search of the most toxic species from mature amyloid fibrils to soluble pre-fibrillar oligomers (dimers to 24-mers).Citation34-Citation39 The formation of structurally distinct amyloid aggregates that share a common “cross-β amyloid” conformation is nowadays proposed to be an inherent property of all proteins,Citation40 probably representing a prominent fold early in the evolution of life.Citation41 Several organisms have taken advantage of the amyloid fold by building functional amyloid structures.Citation42 In this context, mature amyloid aggregates are discussed to have a cytoprotective function by sequestering the soluble toxic oligomeric species that occur on the pathway of protein misfolding.Citation43,Citation44 Thus, prevention of the amyloid formation or solubilization of already formed amyloids may not be a valuable therapeutic strategy to treat protein misfolding diseases,Citation44 in contrast to previous suggestions.Citation45 The greater toxicities of oligomers are assumed to be mediated by their unique structural features,Citation46 but high-resolution knowledge of oligomer structures as well as a molecular understanding of their formation pathways is still lacking.
A large number of studies now indicate that oxidative stress is a suitable trigger for the structural destabilization of the cellular isoform of PrP, which might represent the initial event in the misfolding cascade.Citation13-Citation15,Citation18,Citation19,Citation21,Citation27 In vitro, we and others previously showed that the extend of structural damage determines the population and stability of transitional folding intermediates that direct the misfolding pathway into PrP-specific oligomerization and/or amyloid formation as well as unspecific aggregation followed by precipitation.Citation18,Citation19,Citation21,Citation27 However, it is not clear if the oxidative posttranslational modification, particularly of Met residues, is the causative event that induces the misfolding cascade also in vivo. In this study we designed, produced, and investigated new pseudosulfoxidation mutants of human PrP 121–231 to analyze the impact of methionine oxidation on the structural destabilization as well as the consequences for the conformational transition of PrP in further detail.
Our investigation showed that pseudosulfoxidation of all surface exposed Met residues alters the native PrP fold. The v-hPrP121–231 mutant formed a stable, but partially unfolded state, characterized by a loss in α-helix content. It is questionable if this mutant resembles a specific folding state on the conversion pathway of PrP, or just a conformational burden. However, the even more extended simultaneous pseudosulfoxidation of all methionines in recombinant human PrP by replacement against the highly hydrophilic isosteric analog methoxinine (Mox) recently induced the formation of a β-sheet enriched PrP monomer with strong pro-aggregation behavior, which was suggested to be on the conversion pathway.Citation18 Unfortunately, the sequence of Met oxidation in vivo is not known so far. Differences in the oxidation state of PrPC and PrPSc brain isolates are restricted to the buried helix-3 methionines Met205/Met206 and the partially buried Met213.Citation13-Citation15 This could be explained by the in vivo action of methionine reductases at the accessible Met sites,Citation47 preventing the observation of a stepwise native PrP oxidation that is suggested to depend on the susceptibility of the individual Met residues within their local chemical environment. Except of the three (partially) buried methionines M205/M206/M213, all other surface exposed Met residues have not been associated with detectable structural changes as a consequence of their individual oxidation.Citation17 Thus, the v-hPrP121–231 mutant is expected to combine the conformational rearrangements already known for a single M213S substitution with the accumulated small effects induced by a polarity increase at M129, M134, M154, and M166, if any, mimicking the partially oxidized state before the buried Met residues within the helix-3 region become accessible to oxidation.
Indeed, the overall fold of the v-hPrP121–231 mutant shows striking similarities to monomeric molten globule species of PrP that were recently detected as a consequence of mild copper-catalyzed oxidation and of misfolding under acidic conditions.Citation19,Citation48 Consistently, the basic helical topology remained unchanged but in a less compact form. A gain in β-sheet conformation, as reported for the methoxinine variant of full-length hPrP,Citation18 was observed as a consequence of additional structural perturbation, highlighting the proposed role of Met205 and/or Met206 oxidation as a molecular switch for PrP conversion.Citation17-Citation19 The initial unfolding process mimicked in our study by pseudosulfoxidation of the surface accessible methionines is suggested to slightly reposition the buried M205 and M206 into an oxidation accessible state, finally inducing the structural conversion. This is further supported by the recent observation that oxidation of Met205 and Met212 significantly perturbs the helix-3 interaction network within the hydrophobic core of mouse PrP.Citation19 Moreover, the well-defined oligomeric state detected in this study after metal-induced oxidation of v-hPrP121–231 shares a comparable size and stability with that reported by other in vitro studies applying different triggers for structural conversion.Citation19,Citation21,Citation27,Citation28,Citation49
The individual methionine oxidation at the polymorphic site Met129 does not induce any significant impairment of the consistent conformational properties reported for the α-isoforms of polymorphic Met/Val PrPs,Citation26 as revealed by the site-specific pseudosulfoxidation mutant hPrP121–231 M129T. Met129 is located in a chain region identified as an amyloid nucleating site.Citation50 If an adjacent charge-stabilized hydrogen bond is broken, the N-terminus of hamster PrP is pulled toward the existing β-sheet, which is suggested to promote an initial step within the conversion process.Citation51 However, the individual contribution of a single pseudosulfoxidation at this position was obviously not sufficient to significantly perturb the overall PrP structure.
Compared with wt hPrP121–231 both pseudosulfoxidation mutants show slower kinetics for structural conversion during metal-induced oxidation than expected, considering that these species should mimic already in part oxidized PrP molecules. This gain in stability that clearly depends on the extend of pseudosulfoxidation is mainly attributed to the disparity of the chemical groups introduced by real and pseudosulfoxidation.Citation9 Both Ser and Thr are accepted to be suitable mimics for the associated polarity increase, but do not provide the same sterical demands as methionine sulfoxide. Thus, pseudosulfoxidation most likely resulted in a slightly less destabilized folding state more up in the conversion pathway than obtained by native Met oxidation, associated with a reduced accessibility of the buried residues M205/M206 for ROS, with is suggested to decelerate the subsequent conversion process. Particularly for v-hPrP121–231 we cannot rule out that the simultaneous pseudosulfoxidation created a conformation that is slightly different to that of natively oxidized PrP. However, since the conversion rate is already significantly reduced for the single M129T mutant, the slower conversion kinetics appear to be more likely a result of the disparity of the chemical groups.
Metal-induced oxidation of wt hPrP121–231 results in the formation of highly heterogeneous, unspecific aggregates. The aggregation rate is significantly increased compared with that of hPrP90–231, which favors PrP specific conversion and oligomerization, as previously shown.Citation21 A similar result was obtained monitoring the UV-induced oxidation of the same PrPs.Citation27 These differences are attributed to the ROS quenching capacity located at Met102, Met112, His106, and His111 within the unstructured N-terminal part that is present in hPrP90–231.Citation12 The sacrificial oxidation of these residues is suggested to reduce the level of oxidative stress that affects the folded C-terminal domain of hPrP90–231. This stabilizing effect could be enhanced by a recently proposed direct interaction of the structured part with the N-terminal domain of PrP that was so far considered to be highly flexible,Citation52 as well as by oxidation of other sensitive residues, e.g., tyrosines.Citation21,Citation27 In this context, v-hPrP121–231 has sacrificed more of the ROS quenching capacity than its wild-type counterpart, without increasing its aggregation rate, highlighting again the specific impact of methionine oxidation at surface exposed residues for PrP misfolding.
Taken together, our study indicates that v-hPrP121–231 resembles a specific folding state on the conversion pathway of PrP. In continuation of previous pseudosulfoxidation studies we show that the individual contribution of M129 oxidation to the structural destabilization is not significant. Since M213 is the only methionine accessible for ROS that is known to partly perturb the PrP structure following oxidation, this PrP residue is suggested to be mainly responsible for repositioning the buried M205 and M206 residues into an oxidation accessible state. Particularly the similarities of v-hPrP121–231 to other monomeric molten globule species and its PrP-like aggregation characteristics further strengthens the suggestion that a common misfolding pathway may be shared, if the PrP structure is destabilized by different triggers.Citation19 This is supported by the recent detection of methionine oxidation in the yeast prion Sup35, suggesting an impact of oxidative stress also in the conversion of soluble Sup35 into the aggregated PSI+ isoform,Citation53 as observed for a number of other protein misfolding diseases, including Alzheimer and Parkinson disease.Citation54,Citation55
Materials and Methods
Cloning and gene mutation
The DNA fragment coding for the C-terminal domain of human PrP spanning residues 121–231 (wt hPrP121–231) was amplified by PCR from a plasmid that contains the cDNA of hPrP90–231 and cloned into the prokaryotic expression vector pRSETA containing an N-terminal 6-fold His tag (Invitrogen). For the M129T substitution, a site-directed mutagenesis kit (Stratagene) was applied, while the replacement of the entire surface exposed Met residues (M129, M134, M154, M166, and M213) by serine residues was performed by site-directed mutagenesis of non-overlap extension PCR. Correct mutagenesis and insertion of the PrP coding DNA was verified by automated DNA sequencing.
Protein expression and purification
The generated plasmids were transformed into Escherichia coli BL21 (DE3) pLysS cells (Invitrogen). Protein expression and purification were performed as previously described.Citation21 However, the time for matrix-assisted folding of v-hPrP121–231 was increased by 2.5-fold to yield a soluble conformation. The homogeneity of the purified proteins and the correct folding was verified by SDS-PAGE and CD-spectroscopy, respectively. Protein concentrations were determined by absorption at a wavelength of 280 nm using the molar extinction coefficients ε280 = 16.515 M−1 cm−1 for wt-hPrP121–231 and its mutants.
Oxidative-induced conversion of prion proteins
Structural destabilization and conversion of wild type human PrP121–231 and its mutants into misfolded isoforms was performed by metal-induced oxidation as previously described.Citation21 In brief, PrP aliquots (44 µM) in 10 mM sodium acetate buffer (pH 5.0) were aerobically incubated in the presence of solid copper pellets (16 mg in 60 µl solution) at 37 °C. Due to the oxidation of copper, ROS are continuously formed in solution during incubation, mimicking increasing oxidative stress, as established and validated in Redecke et al. (2007).Citation21 After different time periods the copper pellets were removed to stop the redox reaction and the soluble monomeric and oligomeric PrP isoforms were separated from the highly aggregated and precipitated proteins by centrifugation for 45 min at 20 800 g. The remaining protein concentrations in the supernatants were determined by Bradford assay to calculate the aggregation rate. The obtained values were referred to control samples that did not contain copper pellets.
Circular dichroism (CD) spectroscopy
Far-UV (190–260 nm) CD-spectra were recorded using a Jasco J-715 spectropolarimeter (Jasco, Germany) equipped with a temperature-regulated sample chamber operating at 20 °C in a 1 mm optical pathlength quartz cuvette. Typically, a scanning rate of 100 nm/min and a step resolution of 1 nm were applied. Prior to CD measurements sample solutions were adjusted to a protein concentration of 7.6 µM in 5 mM sodium acetate buffer (pH 5.0). For each measurement five individual scans were averaged, the background spectrum of the corresponding buffer was subtracted, and the data were finally transformed into molar ellipticities (Θ).
Dynamic light scattering (DLS) measurements
DLS measurements were performed using the Spectroscatter 201 (Molecular Dimension, UK) containing a helium-neon laser that operates at a wavelength of λ = 690 nm and a laser power of 10–50 mW. All measurements have been performed at a constant temperature of 20 °C and a scattering angle of 90° using 40 µl sample volumes in a quartz cuvette. An accumulation of 20 measurements per sample was recorded by using the autopilot function. The theoretic hydrodynamic radii and the molecular masses of both the monomeric and the aggregated prion proteins were calculated as previously described.Citation56
| Abbreviations: | ||
| CD | = | circular dichroism |
| CJD | = | Creutzfeldt-Jakob disease |
| DLS | = | dynamic light scattering |
| His | = | histidine |
| Met or M | = | methionine |
| PCR | = | polymerase chain reaction |
| PK | = | Proteinase K |
| Prnp0/0 | = | mice lacking prion protein gene |
| PrP | = | prion protein |
| PrPC | = | cellular prion protein |
| hPrP | = | human prion protein |
| PrPSc | = | infectious isoform of prion protein |
| ROS | = | reactive oxygen species |
| Ser or S | = | serine |
| Thr or T | = | threonine |
| wt | = | wild type |
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
This work was supported by the German Academic Exchange Service (DAAD) (to Elmallah MIY). Redecke L and Betzel C thank the German Federal Ministry for Education and Research (BMBF) for support via grants 01KX0806 and 01KX0807. Financial support from the Hamburg Ministry of Science and Research and Joachim Herz Stiftung as part of the Hamburg Initiative for Excellence in Research (LEXI) and the Hamburg School for Structure and Dynamics (SDI) is gratefully acknowledged.
References
- Weissmann C. The Ninth Datta Lecture. Molecular biology of transmissible spongiform encephalopathies. FEBS Lett 1996; 389:3 - 11; http://dx.doi.org/10.1016/0014-5793(96)00610-2; PMID: 8682199
- Zahn R, Liu A, Lührs T, Riek R, von Schroetter C, López García F, Billeter M, Calzolai L, Wider G, Wüthrich K. NMR solution structure of the human prion protein. Proc Natl Acad Sci U S A 2000; 97:145 - 50; http://dx.doi.org/10.1073/pnas.97.1.145; PMID: 10618385
- Pan KM, Baldwin M, Nguyen J, Gasset M, Serban A, Groth D, Mehlhorn I, Huang Z, Fletterick RJ, Cohen FE, et al. Conversion of alpha-helices into beta-sheets features in the formation of the scrapie prion proteins. Proc Natl Acad Sci U S A 1993; 90:10962 - 6; PMID: 7902575
- Prusiner SB. Novel proteinaceous infectious particles cause scrapie. Science 1982; 216:136 - 44; PMID: 6801762
- Prusiner SB. Molecular biology of prion diseases. Science 1991; 252:1515 - 22; http://dx.doi.org/10.1126/science.1675487; PMID: 1675487
- Weissmann C, Aguzzi A. Bovine spongiform encephalopathy and early onset variant Creutzfeldt-Jakob disease. Curr Opin Neurobiol 1997; 7:695 - 700; http://dx.doi.org/10.1016/S0959-4388(97)80091-8; PMID: 9384548
- Milhavet O, Lehmann S. Oxidative stress and the prion protein in transmissible spongiform encephalopathies. Brain Res Brain Res Rev 2002; 38:328 - 39; http://dx.doi.org/10.1016/S0165-0173(01)00150-3; PMID: 11890980
- Brown DR. Neurodegeneration and oxidative stress: prion disease results from loss of antioxidant defence. Folia Neuropathol 2005; 43:229 - 43; PMID: 16416388
- Stadtman ER. Protein oxidation in aging and age-related diseases. Ann N Y Acad Sci 2001; 928:22 - 38; http://dx.doi.org/10.1111/j.1749-6632.2001.tb05632.x; PMID: 11795513
- Moskovitz J, Bar-Noy S, Williams WM, Requena J, Berlett BS, Stadtman ER. Methionine sulfoxide reductase (MsrA) is a regulator of antioxidant defense and lifespan in mammals. Proc Natl Acad Sci U S A 2001; 98:12920 - 5; PMID: 11606777
- Cui ZJ, Han ZQ, Li ZY. Modulating protein activity and cellular function by methionine residue oxidation. Amino Acids 2012; 43:505 - 17; PMID: 22146868
- Nadal RC, Abdelraheim SR, Brazier MW, Rigby SE, Brown DR, Viles JH. Prion protein does not redox-silence Cu2+, but is a sacrificial quencher of hydroxyl radicals. Free Radic Biol Med 2007; 42:79 - 89; http://dx.doi.org/10.1016/j.freeradbiomed.2006.09.019; PMID: 17157195
- Canello T, Engelstein R, Moshel O, Xanthopoulos K, Juanes ME, Langeveld J, Sklaviadis T, Gasset M, Gabizon R. Methionine sulfoxides on PrPSc: a prion-specific covalent signature. Biochemistry 2008; 47:8866 - 73; http://dx.doi.org/10.1021/bi800801f; PMID: 18680312
- Silva CJ, Onisko BC, Dynin I, Erickson ML, Vensel WH, Requena JR, Antaki EM, Carter JM. Assessing the role of oxidized methionine at position 213 in the formation of prions in hamsters. Biochemistry 2010; 49:1854 - 61; http://dx.doi.org/10.1021/bi901850n; PMID: 20121218
- Colombo G, Meli M, Morra G, Gabizon R, Gasset M. Methionine sulfoxides on prion protein Helix-3 switch on the α-fold destabilization required for conversion. PLoS One 2009; 4:e4296; http://dx.doi.org/10.1371/journal.pone.0004296; PMID: 19172188
- DeMarco ML, Daggett V. Local environmental effects on the structure of the prion protein. C R Biol 2005; 328:847 - 62; http://dx.doi.org/10.1016/j.crvi.2005.05.001; PMID: 16286076
- Lisa S, Meli M, Cabello G, Gabizon R, Colombo G, Gasset M. The structural intolerance of the PrP α-fold for polar substitution of the helix-3 methionines. Cell Mol Life Sci 2010; 67:2825 - 38; http://dx.doi.org/10.1007/s00018-010-0363-1; PMID: 20454997
- Wolschner C, Giese A, Kretzschmar HA, Huber R, Moroder L, Budisa N. Design of anti- and pro-aggregation variants to assess the effects of methionine oxidation in human prion protein. Proc Natl Acad Sci U S A 2009; 106:7756 - 61; http://dx.doi.org/10.1073/pnas.0902688106; PMID: 19416900
- Younan ND, Nadal RC, Davies P, Brown DR, Viles JH. Methionine oxidation perturbs the structural core of the prion protein and suggests a generic misfolding pathway. J Biol Chem 2012; 287:28263 - 75; PMID: 22654104
- Hart T, Hosszu LL, Trevitt CR, Jackson GS, Waltho JP, Collinge J, Clarke AR. Folding kinetics of the human prion protein probed by temperature jump. Proc Natl Acad Sci U S A 2009; 106:5651 - 6; http://dx.doi.org/10.1073/pnas.0811457106; PMID: 19321423
- Redecke L, von Bergen M, Clos J, Konarev PV, Svergun DI, Fittschen UE, Broekaert JA, Bruns O, Georgieva D, Mandelkow E, et al. Structural characterization of β-sheeted oligomers formed on the pathway of oxidative prion protein aggregation in vitro.. J Struct Biol 2007; 157:308 - 20; PMID: 17023178
- Requena JR, Dimitrova MN, Legname G, Teijeira S, Prusiner SB, Levine RL. Oxidation of methionine residues in the prion protein by hydrogen peroxide. Arch Biochem Biophys 2004; 432:188 - 95; http://dx.doi.org/10.1016/j.abb.2004.09.012; PMID: 15542057
- Wong BS, Wang H, Brown DR, Jones IM. Selective oxidation of methionine residues in prion proteins. Biochem Biophys Res Commun 1999; 259:352 - 5; PMID: 10362513
- Abdelraheim SR, Královicová S, Brown DR. Hydrogen peroxide cleavage of the prion protein generates a fragment able to initiate polymerisation of full length prion protein. Int J Biochem Cell Biol 2006; 38:1429 - 40; http://dx.doi.org/10.1016/j.biocel.2006.02.007; PMID: 16595185
- Nyström S, Mishra R, Hornemann S, Aguzzi A, Nilsson KPR, Hammarström P. Multiple substitutions of methionine 129 in human prion protein reveal its importance in the amyloid fibrillation pathway. J Biol Chem 2012; 287:25975 - 84; http://dx.doi.org/10.1074/jbc.M112.372136; PMID: 22669942
- Windl O, Dempster M, Estibeiro JP, Lathe R, de Silva R, Esmonde T, Will R, Springbett A, Campbell TA, Sidle KC, et al. Genetic basis of Creutzfeldt-Jakob disease in the United Kingdom: a systematic analysis of predisposing mutations and allelic variation in the PRNP gene. Hum Genet 1996; 98:259 - 64; http://dx.doi.org/10.1007/s004390050204; PMID: 8707291
- Redecke L, Binder S, Elmallah MIY, Broadbent R, Tilkorn C, Schulz B, May P, Goos A, Eich A, Rübhausen M, et al. UV-light-induced conversion and aggregation of prion proteins. Free Radic Biol Med 2009; 46:1353 - 61; http://dx.doi.org/10.1016/j.freeradbiomed.2009.02.013; PMID: 19249347
- Rezaei H, Eghiaian F, Perez J, Doublet B, Choiset Y, Haertle T, Grosclaude J. Sequential generation of two structurally distinct ovine prion protein soluble oligomers displaying different biochemical reactivities. J Mol Biol 2005; 347:665 - 79; http://dx.doi.org/10.1016/j.jmb.2005.01.043; PMID: 15755458
- Jeganathan S, Hascher A, Chinnathambi S, Biernat J, Mandelkow EM, Mandelkow E. Proline-directed pseudo-phosphorylation at AT8 and PHF1 epitopes induces a compaction of the paperclip folding of Tau and generates a pathological (MC-1) conformation. J Biol Chem 2008; 283:32066 - 76; PMID: 18725412
- Nyström S, Mishra R, Hornemann S, Aguzzi A, Nilsson KPR, Hammarström P. Multiple substitutions of methionine 129 in human prion protein reveal its importance in the amyloid fibrillation pathway. J Biol Chem 2012; 287:25975 - 84; PMID: 22669942
- Prusiner SB, McKinley MP, Bowman KA, Bolton DC, Bendheim PE, Groth DF, Glenner GG. Scrapie prions aggregate to form amyloid-like birefringent rods. Cell 1983; 35:349 - 58; PMID: 6418385
- Rehders D, Claasen B, Redecke L, Buschke A, Reibe C, Jehmlich N, von Bergen M, Betzel C, Meyer B. Peptide NMHRYPNQ of the cellular prion protein (PrP(C)) inhibits aggregation and is a potential key for understanding prion-prion interactions. J Mol Biol 2009; 392:198 - 207; http://dx.doi.org/10.1016/j.jmb.2009.07.014; PMID: 19607841
- Caughey B, Lansbury PT. Protofibrils, pores, fibrils, and neurodegeneration: separating the responsible protein aggregates from the innocent bystanders. Annu Rev Neurosci 2003; 26:267 - 98; http://dx.doi.org/10.1146/annurev.neuro.26.010302.081142; PMID: 12704221
- Simoneau S, Rezaei H, Salès N, Kaiser-Schulz G, Lefebvre-Roque M, Vidal C, Fournier JG, Comte J, Wopfner F, Grosclaude J, et al. In vitro and in vivo neurotoxicity of prion protein oligomers. PLoS Pathog 2007; 3:e125; http://dx.doi.org/10.1371/journal.ppat.0030125; PMID: 17784787
- Walsh DM, Selkoe DJ. Oligomers on the brain: the emerging role of soluble protein aggregates in neurodegeneration. Protein Pept Lett 2004; 11:213 - 28; http://dx.doi.org/10.2174/0929866043407174; PMID: 15182223
- Kayed R, Head E, Thompson JL, McIntire TM, Milton SC, Cotman CW, Glabe CG. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science 2003; 300:486 - 9; http://dx.doi.org/10.1126/science.1079469; PMID: 12702875
- Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, Rowan MJ, Selkoe DJ. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature 2002; 416:535 - 9; http://dx.doi.org/10.1038/416535a; PMID: 11932745
- Winner B, Jappelli R, Maji SK, Desplats PA, Boyer L, Aigner S, Hetzer C, Loher T, Vilar M, Campioni S, et al. In vivo demonstration that alpha-synuclein oligomers are toxic. Proc Natl Acad Sci U S A 2011; 108:4194 - 9; http://dx.doi.org/10.1073/pnas.1100976108; PMID: 21325059
- Silveira JR, Raymond GJ, Hughson AG, Race RE, Sim VL, Hayes SF, Caughey B. The most infectious prion protein particles. Nature 2005; 437:257 - 61; http://dx.doi.org/10.1038/nature03989; PMID: 16148934
- Dobson CM. Protein folding and misfolding. Nature 2003; 426:884 - 90; http://dx.doi.org/10.1038/nature02261; PMID: 14685248
- Chernoff YO. Amyloidogenic domains, prions and structural inheritance: rudiments of early life or recent acquisition?. Curr Opin Chem Biol 2004; 8:665 - 71; http://dx.doi.org/10.1016/j.cbpa.2004.09.002; PMID: 15556413
- Eichner T, Radford SE. A diversity of assembly mechanisms of a generic amyloid fold. Mol Cell 2011; 43:8 - 18; http://dx.doi.org/10.1016/j.molcel.2011.05.012; PMID: 21726806
- Wolfe KJ, Cyr DM. Amyloid in neurodegenerative diseases: friend or foe?. Semin Cell Dev Biol 2011; 22:476 - 81; http://dx.doi.org/10.1016/j.semcdb.2011.03.011; PMID: 21458579
- Duennwald ML. Polyglutamine misfolding in yeast: toxic and protective aggregation. Prion 2011; 5:285 - 90; PMID: 22052348
- Barten DM, Albright CF. Therapeutic strategies for Alzheimer’s disease. Mol Neurobiol 2008; 37:171 - 86; http://dx.doi.org/10.1007/s12035-008-8031-2; PMID: 18581273
- Campioni S, Mannini B, Zampagni M, Pensalfini A, Parrini C, Evangelisti E, Relini A, Stefani M, Dobson CM, Cecchi C, et al. A causative link between the structure of aberrant protein oligomers and their toxicity. Nat Chem Biol 2010; 6:140 - 7; PMID: 20081829
- Binger KJ, Griffin MD, Heinemann SH, Howlett GJ. Methionine-oxidized amyloid fibrils are poor substrates for human methionine sulfoxide reductases A and B2. Biochemistry 2010; 49:2981 - 3; http://dx.doi.org/10.1021/bi902203m; PMID: 20218727
- O’Sullivan DB, Jones CE, Abdelraheim SR, Thompsett AR, Brazier MW, Toms H, Brown DR, Viles JH. NMR characterization of the pH 4 β-intermediate of the prion protein: the N-terminal half of the protein remains unstructured and retains a high degree of flexibility. Biochem J 2007; 401:533 - 40; http://dx.doi.org/10.1042/BJ20060668; PMID: 16958619
- Bocharova OV, Breydo L, Salnikov VV, Baskakov IV. Copper(II) inhibits in vitro conversion of prion protein into amyloid fibrils. Biochemistry 2005; 44:6776 - 87; http://dx.doi.org/10.1021/bi050251q; PMID: 15865423
- Sun Y, Breydo L, Makarava N, Yang Q, Bocharova OV, Baskakov IV. Site-specific conformational studies of prion protein (PrP) amyloid fibrils revealed two cooperative folding domains within amyloid structure. J Biol Chem 2007; 282:9090 - 7; http://dx.doi.org/10.1074/jbc.M608623200; PMID: 17244617
- Alonso DO, DeArmond SJ, Cohen FE, Daggett V. Mapping the early steps in the pH-induced conformational conversion of the prion protein. Proc Natl Acad Sci U S A 2001; 98:2985 - 9; http://dx.doi.org/10.1073/pnas.061555898; PMID: 11248018
- Kaimann T, Metzger S, Kuhlmann K, Brandt B, Birkmann E, Höltje HD, Riesner D. Molecular model of an α-helical prion protein dimer and its monomeric subunits as derived from chemical cross-linking and molecular modeling calculations. J Mol Biol 2008; 376:582 - 96; PMID: 18158160
- Sideri TC, Koloteva-Levine N, Tuite MF, Grant CM. Methionine oxidation of Sup35 protein induces formation of the [PSI+] prion in a yeast peroxiredoxin mutant. J Biol Chem 2011; 286:38924 - 31; PMID: 21832086
- Dong J, Atwood CS, Anderson VE, Siedlak SL, Smith MA, Perry G, Carey PR. Metal binding and oxidation of amyloid-β within isolated senile plaque cores: Raman microscopic evidence. Biochemistry 2003; 42:2768 - 73; http://dx.doi.org/10.1021/bi0272151; PMID: 12627941
- Yamin G, Glaser CB, Uversky VN, Fink AL. Certain metals trigger fibrillation of methionine-oxidized α-synuclein. J Biol Chem 2003; 278:27630 - 5; http://dx.doi.org/10.1074/jbc.M303302200; PMID: 12754258
- Georgieva D, Koker M, Redecke L, Perbandt M, Clos J, Bredehorst R, Genov N, Betzel C. Oligomerization of the proteolytic products is an intrinsic property of prion proteins. Biochem Biophys Res Commun 2004; 323:1278 - 86; http://dx.doi.org/10.1016/j.bbrc.2004.08.230; PMID: 15451435