Abstract
The pathogenic mechanism of prion diseases remains unknown. We recently reported that prion infection disturbs post-Golgi trafficking of certain types of membrane proteins to the cell surface, resulting in reduced surface expression of membrane proteins and abrogating the signal from the proteins. The surface expression of the membrane proteins was reduced in the brains of mice inoculated with prions, well before abnormal symptoms became evident. Prions or pathogenic prion proteins were mainly detected in endosomal compartments, being particularly abundant in recycling endosomes. Some newly synthesized membrane proteins are delivered to the surface from the Golgi apparatus through recycling endosomes, and some endocytosed membrane proteins are delivered back to the surface through recycling endosomes. These results suggest that prions might cause neuronal dysfunctions and cell loss by disturbing post-Golgi trafficking of membrane proteins via accumulation in recycling endosomes. Interestingly, it was recently shown that delivery of a calcium channel protein to the cell surface was impaired and its function was abrogated in a mouse model of hereditary prion disease. Taken together, these results suggest that impaired delivery of membrane proteins to the cell surface is a common pathogenic event in acquired and hereditary prion diseases.
Introduction
The normal cellular prion protein, designated PrPC, is a glycosylphosphatidylinositol (GPI)-anchored cell surface glycoprotein most abundantly expressed by neurons and to a lesser extent in other tissues such as lymph nodes and spleen.Citation1 Conformational conversion of PrPC into the abnormally folded, amyloidogenic isoform, PrPSc, is a central event in the pathogenesis of prion diseases, which include Creutzfeldt-Jacob disease (CJD) in humans and scrapie and bovine spongiform encephalopathy in animals.Citation1 Indeed, mice devoid of PrPC (Prnp0/0) are resistant to prions, neither producing PrPSc nor developing prion disease even after intracerebral inoculation with the prions.Citation2-Citation5 However, the pathogenic mechanism underlying neuronal cell death in prion diseases remains largely unknown.
Cell surface expression levels of membrane proteins are tightly controlled via complicated vesicular transport systems. Newly synthesized membrane proteins are delivered from the endoplasmic reticulum (ER) to the Golgi apparatus, where sugar chains attach to the proteins, and then to the cell surface directly or indirectly via recycling endosome compartments.Citation6,Citation7 The membrane proteins expressed on the surface are endocytosed to sorting endosome compartments, with some of them being delivered back to the cell surface directly or indirectly via recycling endosome compartments and others are transported to lysosomes for degradation.Citation6,Citation7 Damage to the vesicular transport of membrane proteins would reduce surface expression levels of the membrane molecules, leading to functional deficiency of the molecules eventually causing damage to the cells.
We recently reported that prion infection disturbs post-Golgi vesicular trafficking and hinders membrane proteins from being delivered to the cell surface from the Golgi apparatus.Citation8 These findings suggest that disturbance of post-Golgi vesicular trafficking is a pathogenic mechanism in prion diseases.
Prion Infection Impairs post-Golgi Vesicular Trafficking
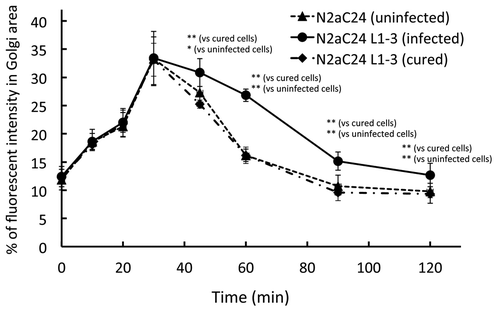
A vesicular transport assay using a temperature-sensitive mutant of the vesicular stomatitis virus-G protein fused with green fluorescent protein, designated VSV-G(ts045)-GFP, is very useful to assess vesicular trafficking from the ER to the cell surface through the Golgi apparatus in cells.Citation9 VSV-G(ts045)-GFP folds improperly at non-permissive temperature, thereby remaining in the ER. In contrast, at permissive temperature, this protein is properly folded and exits from the ER to the Golgi apparatus and then to the cell surface. VSV-G(ts045)-GFP was normally transported from the ER to the Golgi apparatus in 22L prion-infected N2aC24L1–3 cells at the permissive temperature, compared with uninfected N2aC24 cells ().Citation8 However, the export of VSV-G(ts045)-GFP from the Golgi apparatus was significantly delayed in infected cells, and this delayed export of VSV-G(ts045)-GFP was recovered to a normal level in cured N2aC24L1–3 cells, in which prions had been completely eliminated by treatment with SAF32 anti-PrP antibody ().Citation8 Similar delayed export of VSV-G(ts045)-GFP from the Golgi apparatus was observed in Chandler prion-infected N2aC24Chm cells.Citation8 This delayed export was also recovered to a normal level in cured N2aC24Chm cells.Citation8 These results indicate that prion infection disturbs post-Golgi vesicular trafficking, but does not affect vesicular trafficking from the ER to the Golgi apparatus.
Figure 1. VSV-G(ts045)-GFP transport assay in uninfected N2aC24, infected N2aC24L1–3, and cured N2aC24L1–3 cells. Cells were transfected with the vector encoding VSV-G(ts045)-GFP and incubated at non-permissive temperature. Fluorescent intensities for VSV-G(ts045)-GFP at the Golgi region against those in the whole cell were determined in the randomly selected transfected cells (n = 14–16) at various times after the cells were transferred to permissive temperature. The Golgi area was immunohistochemically delineated using the Golgi marker GM130. Trafficking of VSV-G(ts045)-GFP in the cells was assessed as the accumulation kinetics of the protein in the Golgi area. Post-Golgi trafficking of VSV-G(ts045) was significantly delayed in infected N2aC24L1–3 cells, compared with that in uninfected N2aC24 cells. No significant difference in the VSV-G(ts045)-GFP trafficking was observed between uninfected N2aC24 and cured N2aC24L1–3 cells. Data were analyzed using the Student t test. * P < 0.05, ** P < 0.01. These were modified from Uchiyama et al.Citation8
Prion Infection Selectively Impairs Trafficking of Membrane Proteins to the Cell Surface
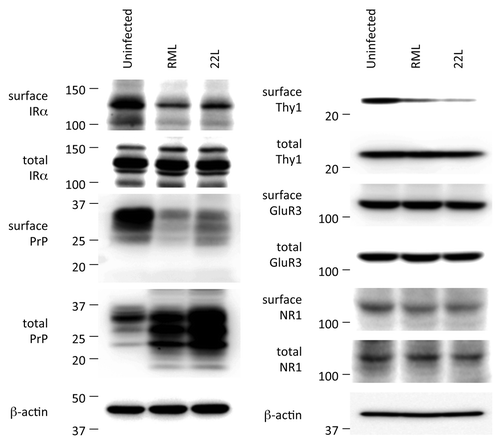
To investigate the consequences of the disturbed post-Golgi vesicular trafficking, surface membrane proteins in uninfected N2aC24, infected N2aC24L1–3, and cured N2aC24L1–3 cells were labeled with biotins and subjected to western blotting after purification of the biotinylated proteins. Three membrane proteins we examined, including PrP, the insulin receptor α subunit (IRα) and attractin, whose mutation causes prion disease-like spongiform neurodegeneration in animals,Citation10 were significantly less biotinylated in infected cells than in uninfected and cured cells,Citation8 indicating that surface expression of these membrane proteins is lower in infected cells than in uninfected and cured cells. Less than 1% of the biotinylated PrP from infected cells were PK-resistant,Citation8 indicating that most of the biotinylated PrP in infected cells are PrPC. Total proteins (unbiotinylated and biotinylated) composed of IRα and attractin were the same in these cells although, due to accumulation of PrPSc, total PrP was higher in infected cells than in uninfected and cured cells.Citation8 Consistent with these results, the cell surface staining of PrPC and IRα was reduced in infected cells.Citation8 Instead, these molecules were aberrantly accumulated at a paranuclear region, most parts of which were co-stained with the Golgi markers TGN38 and giantin.Citation8 Uninfected and cured cells exhibited staining for PrPC and IRα predominantly on the cell surface. PrPC, IRα and another GPI-anchored protein, Thy-1, were also less biotinylated in brain slices from RML prion- and 22L prion-infected, terminally ill mice than in control uninfected brain slices ().Citation8 However, the glutamate receptor subunits GluR3 and NR1 were similarly biotinylated in infected and uninfected brain slices ().Citation8 PrPC and Thy-1 are GPI-anchored molecules, IR and attractin are single-pass transmembrane molecules, and GluR3 and NR1 are multipass transmembrane molecules. These results indicate that prion infection might impair post-Golgi trafficking of certain types of membrane proteins.
Figure 2. Western blotting of surface and total membrane proteins in the brains of uninfected and infected mice. Brains were freshly removed from uninfected control and terminally ill mice and coronally sliced. The brain slices were then subjected to biotin labeling of membrane proteins. The biotin-labeled surface proteins were purified using avidin-beads and subjected to western blotting for detection of IRα, PrP, Thy-1, GluR3, and NR1. Total expression levels of each protein were also determined. Biotinylated levels of IRα, PrP, and Thy-1, but not GluR3 and NR1, were lower in infected brains than in uninfected brains. Total expression levels of each protein were the same except for PrP. Total PrP was increased in infected brains due to accumulation of PrPSc. β-actin is an internal control. These were modified from Uchiyama et al.Citation8
Trafficking of Membrane Proteins Is Commonly Impaired in Prion Diseases
Prion diseases in humans manifest as sporadic, hereditary, and acquired types. Hereditary prion diseases including familial CJD and fatal familial insomnia (FFI) are causatively associated with specific mutations of the PrP gene. Interestingly, reduced surface expression was also shown for GPI-anchored GFP in N2a cells expressing the FFI-linked PrP mutant, D177N/Met-128 PrP,Citation11 and for the voltage-gated calcium channel α2δ-1 subunit in the primary cerebellar granule neurons from transgenic mice expressing familial CJD-associated PrP mutants, PG14 PrP and D177N/Val-128.Citation12 Taken together with our results obtained from prion-infected cells and mice,Citation8 trafficking of membrane proteins to the cell surface could be commonly impaired in acquired and hereditary prion diseases.
Disturbed Trafficking of Membrane Proteins Is an Early Event in Prion Diseases
To investigate the role of the disturbed post-Golgi vesicular trafficking in the pathogenesis of prion disease, PrPC and IRα were biotin-labeled in brain slices from mice sacrificed at 2, 62 to 68, and 80 to 87 days post-inoculation (dpi) with RML prions and at the terminal stage. The infected mice became ill at around 112 dpi. PrPC and IRα were biotinylated at the same level in uninfected and infected brains at 2 dpi.Citation8 However, at 80–87 dpi, they were significantly less biotinylated in infected brains than in uninfected brains.Citation8 They were also less biotinylated in infected brains at 62–68 dpi.Citation8 PrPSc was detectable in the brains from 68 dpi and in an increased amount at 87 dpi, reaching its maximal level at the terminal stage.Citation8 These results suggest that post-Golgi trafficking is disturbed in brains from the early stage of the infection soon after accumulation of PrPSc.
Early impairment of post-Golgi trafficking was also suggested in infected mice transgenic for the GPI-anchored membrane protein PrP-EGFP, a fusion protein of PrP with EGFP.Citation13 Tg mice expressing PrP-EGFP together with wild-type PrPC developed the disease after inoculation with RML prions, with accumulation of PrP-EGFP in the Golgi apparatus in neurons from the early stages after infection.Citation13 It was also reported that impaired delivery of the voltage-gated calcium channel α2δ-1 subunit to the cell surface in Tg(PG14) mice is an early pathogenic event, by demonstrating that glutamate release and calcium influx evoked by K+-induced depolarization in the cerebellar synaptosomes was impaired well before clinical symptoms became evident in Tg(PG14) mice.Citation12 These results suggest that impairment of trafficking of membrane proteins to the surface is a common early pathogenic event in acquired and hereditary prion diseases.
Possible Mechanisms for Impaired Trafficking of Membrane Proteins in Prion Diseases
Subcellular localization of PrPSc was determined in infected N2aC24L1–3 and N2aC24Chm cells using anti-PrP monoclonal Ab mAb132, which is able to specifically visualize PrPSc under partially denatured conditions by binding to residues 119–127 of mouse PrP. PrPSc was abundantly detected at a paranuclear region, which was preferentially costained with the recycling endosome marker transferrin receptor and to a lesser extent with other endosome markers, including another recycling endosome marker Rab11, the early endosome marker EEA1, and the late endosome marker Rab9.Citation8 Only a small proportion of PrPSc were detected in the Golgi apparatus.Citation8 Aberrant accumulation of PrPSc in recycling endosomes has also been reported in other infected culture cells and in neurons of RML prion-infected mice.Citation14,Citation15 These results indicate that PrPSc accumulates throughout endosomal compartments, particularly abundantly in the recycling endosome compartment, but little in the Golgi apparatus. Newly synthesized membrane proteins are delivered from the ER to the Golgi apparatus and then to the cell surface directly or indirectly via recycling endosome compartments.Citation6,Citation7 Moreover, the membrane proteins on the surface are endocytosed to sorting endosome compartments and some of them are delivered back to the cell surface directly or indirectly via recycling endosome compartments.Citation6,Citation7 It is thus possible that PrPSc abnormally accumulated in recycling endosome might affect transport of membrane proteins from the Golgi apparatus to the recycling endosome and then to the plasma membrane. However, D177N/Met-128 mutant PrP was mainly accumulated in the ER and Golgi apparatus of transfected N2a cells and upregulated the Rab GDP dissociation inhibitor α but in contrast, downregulated Rab11, leading to impairment of vesicular trafficking of the GPI-anchored GFP to the cell surface.Citation11 PG14 PrP was also retained in the ER, causing abnormal intracellular accumulation of the voltage-gated calcium channel α2δ-1 subunit via interaction with the subunit.Citation12 These results suggest that trafficking of membrane proteins to the surface is disturbed by different mechanisms in acquired and hereditary prion diseases.
Implications for the Pathogenesis of Prion Disease
We here showed that impaired vesicular trafficking of membrane proteins to the cell surface could be a common early pathogenic mechanism in prion disease. The impaired trafficking of membrane proteins caused reduced surface expression of the proteins in vitro and in vivo, suggesting that these proteins are functionally deficient in infected cells. Indeed, the downstream signal of IR was weakened in infected cells after stimulation with insulin.Citation8 Moreover, it was shown that glutamate release and calcium influx evoked by K+-induced depolarization were impaired in the cerebellar synaptosomes of Tg(PG14) mice due to the decreased surface expression of the voltage-gated calcium channel α2δ-1 subunit.Citation12 It is thus conceivable that prions impair trafficking of membrane proteins to the cell surface and consequently reduce surface expression of membrane proteins, thereby causing functional deficiency of the proteins leading to neuronal dysfunctions and cell death. On the other hand, the disturbed post-Golgi vesicular trafficking caused the aberrant accumulation of membrane proteins in the Golgi apparatus in infected cells.Citation8 It has been reported that abnormal accumulation of membrane or secretory proteins in the Golgi apparatus could induce ER stress.Citation16 It is therefore possible that prion infection might induce ER stress by disturbing post-Golgi vesicular trafficking and consequently causing aberrant accumulation of membrane proteins in the Golgi apparatus. Indeed, elevated ER stress has been reported in prion-infected cells and brains.Citation17,Citation18 Other mechanisms have been proposed to explain cell death in prion diseases. Full-length PrPSc and its fragments were shown to be toxic to primary neurons.Citation17,Citation19 Transmembrane and cytosolic PrPs, termed CtmPrP and CytPrP, respectively, were also suggested to be neurotoxic molecules in prion diseases.Citation20,Citation21 Moreover, impairment of the ubiquitin-proteasome system was demonstrated in prion-infected ScN2a cells and mice.Citation22 Further studies are required to understand the neurotoxic mechanism leading to neuronal cell death in prion diseases.
Similar abnormal phenotypes to those often observed in human prion diseases have been reported in Prnp0/0 mice, such as demyelination in the spinal cord and peripheral nerves,Citation23 alteration of sleep patterns and circadian rhythms,Citation24 and defective memory and learning.Citation25,Citation26 However, spongiform neurodegeneration and neuronal cell loss have not been observed in Prnp0/0 mice.Citation27 It is therefore possible that the reduced surface expression of PrPC that results in functional deficiency of PrPC might be responsible for some aspects of the pathogenesis in prion diseases, such as those observed in Prnp0/0 mice, but not in spongiform degeneration and neuronal cell death. Prion disease-like spongiform neurodegeneration is found in animals lacking functional attractin,Citation10 suggesting that functional deficiency of attractin that is caused by its reduced surface expression might be involved in spongiform degeneration in prion diseases. However, it was recently reported that mice deficient for attractin developed prion disease with the same incubation times as control wild-type mice after inoculation with prions.Citation28 Reduced insulin signaling is suggested to be involved in the pathogenesis of neurodegenerative disorders, such as Alzheimer disease, by reducing its neuroprotective function and dysregulating synaptic plasticity and memory formation.Citation29,Citation30 It remains to be elucidated whether or not insulin signal could be also involved in the pathogenesis of prion disease.
We here discussed that prions could disturb trafficking of certain types of membrane proteins to the cell surface, and that the disturbed trafficking could eventually cause neuronal dysfunctions associated with the pathogenesis of prion disease. Thus, it might be worth elucidating the molecular mechanism underlying the impaired trafficking of membrane proteins to the surface in prion-infected cells for further understanding of the pathogenesis of prion diseases and development of treatments for prion diseases.
| Abbreviations: | ||
| PrPC | = | normal cellular prion protein |
| PrPSc | = | abnormally folded, amyloidgenic prion protein |
| Prnp0/0 mice | = | mice devoid of PrPC |
| GPI | = | glycosylphosphatidylinositol |
| CJD | = | Creutzfeldt-Jacob disease |
| FFI | = | fatal familial insomnia |
| ER | = | endoplasmic reticulum |
| VSV-G(ts045)-GFP | = | temperature-sensitive mutant of the vesicular stomatitis virus-G protein fused with green fluorescent protein |
| IRα | = | insulin receptor α subunit |
| PK | = | proteinase K |
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
This study was partly supported by a Grant-in-Aid from the BSE and other Prion Disease Control Project of the Ministry of Agriculture, Forestry and Fisheries of Japan, Grants-in-Aid from the Research Committee of Prion Disease and Slow Virus infection, the Ministry of Health, Labour and Welfare of Japan, and a Grant-in-Aid for TSE research (H23-Shokuhin-Ippan-005) and Research on Measures for Intractable Diseases from the Ministry of Health, Labour and Welfare of Japan. H.M. was partly supported by a Cooperative Research Grant of the Institute for Enzyme Research, The University of Tokushima.
References
- Prusiner SB. Prions. Proc Natl Acad Sci U S A 1998; 95:13363 - 83; http://dx.doi.org/10.1073/pnas.95.23.13363; PMID: 9811807
- Büeler H, Aguzzi A, Sailer A, Greiner RA, Autenried P, Aguet M, Weissmann C. Mice devoid of PrP are resistant to scrapie. Cell 1993; 73:1339 - 47; http://dx.doi.org/10.1016/0092-8674(93)90360-3; PMID: 8100741
- Prusiner SB, Groth D, Serban A, Koehler R, Foster D, Torchia M, Burton D, Yang SL, DeArmond SJ. Ablation of the prion protein (PrP) gene in mice prevents scrapie and facilitates production of anti-PrP antibodies. Proc Natl Acad Sci U S A 1993; 90:10608 - 12; http://dx.doi.org/10.1073/pnas.90.22.10608; PMID: 7902565
- Manson JC, Clarke AR, McBride PA, McConnell I, Hope J. PrP gene dosage determines the timing but not the final intensity or distribution of lesions in scrapie pathology. Neurodegeneration 1994; 3:331 - 40; PMID: 7842304
- Sakaguchi S, Katamine S, Shigematsu K, Nakatani A, Moriuchi R, Nishida N, Kurokawa K, Nakaoke R, Sato H, Jishage K, et al. Accumulation of proteinase K-resistant prion protein (PrP) is restricted by the expression level of normal PrP in mice inoculated with a mouse-adapted strain of the Creutzfeldt-Jakob disease agent. J Virol 1995; 69:7586 - 92; PMID: 7494265
- Maxfield FR, McGraw TE. Endocytic recycling. Nat Rev Mol Cell Biol 2004; 5:121 - 32; http://dx.doi.org/10.1038/nrm1315; PMID: 15040445
- Hsu VW, Prekeris R. Transport at the recycling endosome. Curr Opin Cell Biol 2010; 22:528 - 34; http://dx.doi.org/10.1016/j.ceb.2010.05.008; PMID: 20541925
- Uchiyama K, Muramatsu N, Yano M, Usui T, Miyata H, Sakaguchi S. Prions disturb post-Golgi trafficking of membrane proteins. Nat Commun 2013; 4:1846; http://dx.doi.org/10.1038/ncomms2873; PMID: 23673631
- Kasai K, Shin HW, Shinotsuka C, Murakami K, Nakayama K. Dynamin II is involved in endocytosis but not in the formation of transport vesicles from the trans-Golgi network. J Biochem 1999; 125:780 - 9; http://dx.doi.org/10.1093/oxfordjournals.jbchem.a022349; PMID: 10101292
- Kuramoto T, Kitada K, Inui T, Sasaki Y, Ito K, Hase T, Kawagachi S, Ogawa Y, Nakao K, Barsh GS, et al. Attractin/mahogany/zitter plays a critical role in myelination of the central nervous system. Proc Natl Acad Sci U S A 2001; 98:559 - 64; http://dx.doi.org/10.1073/pnas.98.2.559; PMID: 11209055
- Massignan T, Biasini E, Lauranzano E, Veglianese P, Pignataro M, Fioriti L, Harris DA, Salmona M, Chiesa R, Bonetto V. Mutant prion protein expression is associated with an alteration of the Rab GDP dissociation inhibitor alpha (GDI)/Rab11 pathway. Mol Cell Proteomics 2010; 9:611 - 22; http://dx.doi.org/10.1074/mcp.M900271-MCP200; PMID: 19996123
- Senatore A, Colleoni S, Verderio C, Restelli E, Morini R, Condliffe SB, Bertani I, Mantovani S, Canovi M, Micotti E, et al. Mutant PrP suppresses glutamatergic neurotransmission in cerebellar granule neurons by impairing membrane delivery of VGCC α(2)δ-1 Subunit. Neuron 2012; 74:300 - 13; http://dx.doi.org/10.1016/j.neuron.2012.02.027; PMID: 22542184
- Barmada SJ, Harris DA. Visualization of prion infection in transgenic mice expressing green fluorescent protein-tagged prion protein. J Neurosci 2005; 25:5824 - 32; http://dx.doi.org/10.1523/JNEUROSCI.1192-05.2005; PMID: 15958749
- Yamasaki T, Suzuki A, Shimizu T, Watarai M, Hasebe R, Horiuchi M. Characterization of intracellular localization of PrP(Sc) in prion-infected cells using a mAb that recognizes the region consisting of aa 119-127 of mouse PrP. J Gen Virol 2012; 93:668 - 80; http://dx.doi.org/10.1099/vir.0.037101-0; PMID: 22090211
- Godsave SF, Wille H, Kujala P, Latawiec D, DeArmond SJ, Serban A, Prusiner SB, Peters PJ. Cryo-immunogold electron microscopy for prions: toward identification of a conversion site. J Neurosci 2008; 28:12489 - 99; http://dx.doi.org/10.1523/JNEUROSCI.4474-08.2008; PMID: 19020041
- Xu YX, Liu L, Caffaro CE, Hirschberg CB. Inhibition of Golgi apparatus glycosylation causes endoplasmic reticulum stress and decreased protein synthesis. J Biol Chem 2010; 285:24600 - 8; http://dx.doi.org/10.1074/jbc.M110.134544; PMID: 20529871
- Hetz C, Russelakis-Carneiro M, Maundrell K, Castilla J, Soto C. Caspase-12 and endoplasmic reticulum stress mediate neurotoxicity of pathological prion protein. EMBO J 2003; 22:5435 - 45; http://dx.doi.org/10.1093/emboj/cdg537; PMID: 14532116
- Hetz C, Russelakis-Carneiro M, Wälchli S, Carboni S, Vial-Knecht E, Maundrell K, Castilla J, Soto C. The disulfide isomerase Grp58 is a protective factor against prion neurotoxicity. J Neurosci 2005; 25:2793 - 802; http://dx.doi.org/10.1523/JNEUROSCI.4090-04.2005; PMID: 15772339
- Forloni G, Angeretti N, Chiesa R, Monzani E, Salmona M, Bugiani O, Tagliavini F. Neurotoxicity of a prion protein fragment. Nature 1993; 362:543 - 6; http://dx.doi.org/10.1038/362543a0; PMID: 8464494
- Hegde RS, Mastrianni JA, Scott MR, DeFea KA, Tremblay P, Torchia M, DeArmond SJ, Prusiner SB, Lingappa VR. A transmembrane form of the prion protein in neurodegenerative disease. Science 1998; 279:827 - 34; http://dx.doi.org/10.1126/science.279.5352.827; PMID: 9452375
- Ma J, Wollmann R, Lindquist S. Neurotoxicity and neurodegeneration when PrP accumulates in the cytosol. Science 2002; 298:1781 - 5; http://dx.doi.org/10.1126/science.1073725; PMID: 12386337
- Kristiansen M, Deriziotis P, Dimcheff DE, Jackson GS, Ovaa H, Naumann H, Clarke AR, van Leeuwen FW, Menéndez-Benito V, Dantuma NP, et al. Disease-associated prion protein oligomers inhibit the 26S proteasome. Mol Cell 2007; 26:175 - 88; http://dx.doi.org/10.1016/j.molcel.2007.04.001; PMID: 17466621
- Nishida N, Tremblay P, Sugimoto T, Shigematsu K, Shirabe S, Petromilli C, Erpel SP, Nakaoke R, Atarashi R, Houtani T, et al. A mouse prion protein transgene rescues mice deficient for the prion protein gene from purkinje cell degeneration and demyelination. Lab Invest 1999; 79:689 - 97; PMID: 10378511
- Tobler I, Gaus SE, Deboer T, Achermann P, Fischer M, Rülicke T, Moser M, Oesch B, McBride PA, Manson JC. Altered circadian activity rhythms and sleep in mice devoid of prion protein. Nature 1996; 380:639 - 42; http://dx.doi.org/10.1038/380639a0; PMID: 8602267
- Collinge J, Whittington MA, Sidle KC, Smith CJ, Palmer MS, Clarke AR, Jefferys JG. Prion protein is necessary for normal synaptic function. Nature 1994; 370:295 - 7; http://dx.doi.org/10.1038/370295a0; PMID: 8035877
- Nishida N, Katamine S, Shigematsu K, Nakatani A, Sakamoto N, Hasegawa S, Nakaoke R, Atarashi R, Kataoka Y, Miyamoto T. Prion protein is necessary for latent learning and long-term memory retention. Cell Mol Neurobiol 1997; 17:537 - 45; http://dx.doi.org/10.1023/A:1026315006619; PMID: 9353594
- Büeler H, Fischer M, Lang Y, Bluethmann H, Lipp HP, DeArmond SJ, Prusiner SB, Aguet M, Weissmann C. Normal development and behaviour of mice lacking the neuronal cell-surface PrP protein. Nature 1992; 356:577 - 82; http://dx.doi.org/10.1038/356577a0; PMID: 1373228
- Gunn TM, Carlson GA. RML prions act through Mahogunin and Attractin-independent pathways. Prion 2013; 7:267 - 71; http://dx.doi.org/10.4161/pri.25054; PMID: 23787699
- Gasparini L, Netzer WJ, Greengard P, Xu H. Does insulin dysfunction play a role in Alzheimer’s disease?. Trends Pharmacol Sci 2002; 23:288 - 93; http://dx.doi.org/10.1016/S0165-6147(02)02037-0; PMID: 12084635
- Nelson TJ, Alkon DL. Insulin and cholesterol pathways in neuronal function, memory and neurodegeneration. Biochem Soc Trans 2005; 33:1033 - 6; http://dx.doi.org/10.1042/BST20051033; PMID: 16246039