
Abstract
Prion and Alzheimer diseases are fatal neurodegenerative diseases caused by misfolding and aggregation of the cellular prion protein (PrPC) and the β-amyloid peptide, respectively. Soluble oligomeric species rather than large aggregates are now believed to be neurotoxic. PrPC undergoes three proteolytic cleavages as part of its natural life cycle, α-cleavage, β-cleavage, and ectodomain shedding. Recent evidences demonstrate that the resulting secreted PrPC molecules might represent natural inhibitors against soluble toxic species. In this mini-review, we summarize recent observations suggesting the potential benefit of using PrPC-derived molecules as therapeutic agents in prion and Alzheimer diseases.
Numerous neurodegenerative diseases including prion and Alzheimer diseases are caused by aberrant folding and aggregation of amyloid proteins into toxic intermediates. Blocking the formation or sequestrating these toxic intermediates may represent an interesting therapeutical avenue to reduce neurodegeneration occurring in these diseases.
Secreted Forms of PrPC in Prion Diseases
Prion diseases are fatal transmissible neurodegenerative disorders caused by the misfolding and aggregation of the cellular prion protein (PrPC) into a pathological isoform partially resistant to proteinase K termed PrPres Citation1. Newly converted PrPres can recruit other PrPC molecules and cause their conversion into more PrPres. The assembly of PrPres molecules form infectious particles termed prions that are responsible for the transmission of prion diseases. The plasma membrane and the endosomal recycling compartment are the primary site for prion conversion.Citation2,Citation3 Prion conversion is central to prion neurotoxicity.Citation4 In humans, most prion diseases are sporadic and occur in the absence of mutation in the cellular prion protein (PrPC) gene, PRNP, and in the absence of any contact with infectious prions. In familial cases, inherited dominant mutations in the PRNP gene cause the production of abnormal PrPC molecules. Iatrogenic and acquired cases constitute a third category of prion diseases and result from accidental exposures to prions
PrPC is translated as a precursor, and the mature protein is a 209 amino acid GPI anchored polypeptide present at the cell surface. PrPC is composed of an unstructured N-terminal domain and a structured C-terminal domain with three α-helices and two short β-sheets.Citation5 Both domain are essential for efficient conversion of PrPC into PrPres Citation6,Citation7. PrPC has a dual function in prion diseases. PrPC is the substrate for prion conversion and its neuronal expression is required for the pathogenesis of prion diseases.Citation4,Citation8,Citation9 In addition, PrPC is a cell surface receptor mediating neurotoxic signals for β-sheet soluble oligomers produced during prion conversion.Citation10,Citation11 In contrast to its role in prion diseases, the physiological function of PrPC is still unclear.
En route to the cell surface, a fraction of PrPC undergoes an endoproteolytic cleavage, termed α-cleavage, between amino acid 110–111 and 112. This cleavage causes the release of PrPN1, a N-terminal moiety of 88 amino acids. The C-terminal counterpart, PrPC1, remains at the cell surface.Citation12,Citation13 In the brain, up to 50% of PrPC molecules undergo the α-cleavage and this physiological cleavage may be considered a major post-translational modification of this protein.Citation13 The identity of the protease responsible for the α-cleavage is still unknown.Citation14 A second physiological cleavage also termed shedding is performed by the metalloprotease ADAM10 at the cell surface and leads to the release of a full-length anchorless PrPC isoform (shed PrP).Citation15-Citation17 PrPC shedding contributes to a significant proportion of secreted PrPC molecules in cell culture and shed PrPC was also detected in vivo.Citation16-Citation19 A third cleavage between amino acid 89 and 90 also termed β-cleavage is activated by ROS and may be involved in the cellular response to oxidative stress.Citation12,Citation20 This modification causes the release of PrPN2, and leaves the C-terminal PrPC2 moiety at the cell surface.
PrPC proteolytic cleavages may have important functions.Citation14,Citation20,Citation21 In particular, they may be considered as endogenous mechanisms of defense against prion disease for several reasons (). First, they reduce the expression of GPI-anchored PrPC which is essential for efficient prion conversion in cell cultures, the development of prion diseases in mice models, and central nervous system invasion after peripheral prion infection.Citation4,Citation8,Citation9,Citation22-Citation24 GPI-anchored PrPC is also a cell surface receptor for prion soluble oligomers and relay intracellular toxic signals.Citation10,Citation11 Releasing PrPC from the cell surface is expected to interfere with these neurotoxic signals. Second, constitutive secretion of PrPC using a genetic fusion between PrPC and immunoglobulin Fc gamma interferes with the propagation of PrPres in experimentally-infected mice and delays onset of disease after experimental inoculation with prions.Citation25 The mechanism involves the capture and neutralization of PrPres. In agreement with this result, recombinant PrP reduces prion conversion in vitro by binding to PrPres and preventing PrPres/PrPC interactions.Citation6 Third, PrPC1 produced after PrPC α-cleavage is not a substrate for prion conversion. In contrast, PrPC1 acts as a dominant-negative inhibitor of PrPres mediated-conversion suggesting that an incomplete PrPC molecule at the cell surface can trap PrPres into a non-convertible complex.Citation26 Fourth, the observation that two disease-associated PrPC mutants do not undergo ROS-mediated β-cleavage suggests that PrPN2 and/or PrPC2 have an important role against oxidative stress in prion diseases.Citation20 In agreement with the protective role of PrPC α-cleavage, a decrease in PrPC1 levels has been observed in animal models of prion diseases and in a human prion disease.Citation13,Citation26,Citation27 This decrease is mediated by an increased activity of 3-phosphoinositide–dependent kinase-1 (PDK1) in prion-infected neurons, and siRNA-mediated silencing of PDK1 delays mortality in prion-infected mice.Citation28 Also, the annual incidence of prion diseases is approximately 1 in 1 000 000 persons.Citation29 Thus, these disorders are among the most rare neurodegeneratives diseases, and physiological proteolytic cleavages of PrPC may in part explain this very low incidence rate.
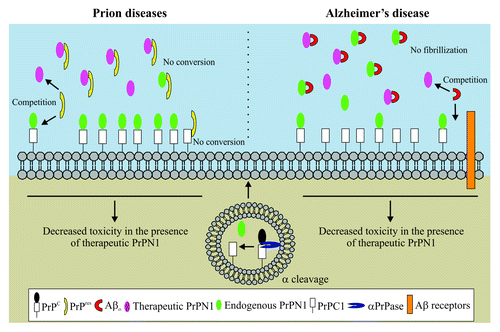
Figure 1. PrPC α-cleavage display hypothetical neuroprotective functions in prion and Alzheimer diseases. In prion diseases (left), endogenously produced PrPN1 (green) traps PrPres toxic species, inhibits their conversion and thus, reduces their toxicity. In addition, PrPC1 is a dominant-negative inhibitor of PrPres formation. In Alzheimer disease (right), PrPC α-cleavage is increased. PrPN1 molecules trap Aβ oligomers (Aβo) and reduce Aβ-mediated toxicity by competition with Aβo receptors at the cell surface. Furthermore, the lack of Aβ binding sites on PrPC1 prevents PrPC1 from mediating Aβo toxicity. In a curative approach, therapeutic PrPN1 or PrPN1-derived drugs (purple) could be administered to supplement endogenous PrPN1 in both prion and Alzheimer diseases.
All these evidences point to two possible therapeutic avenues in prion diseases. As a first strategy, PrPC-derived molecules may be used to capture PrPres, prevent PrPC/PrPres interactions at the cell surface and reduce prion conversion. In addition, it is tempting to take advantage of the physiological cleavages of PrPC to try and activate the involved proteases. We propose that PrPC α-cleavage is the most promising therapeutic target because it removes the substrate for prion conversion. In contrast, too much PrPC shedding may be neurotoxic.Citation30
Secreted Forms of PrPC in Alzheimer Diseases
Alzheimer disease accounts for an estimated 60 to 80% of the current 35 million individuals living with dementia worldwide.Citation31 An important area of research into the molecular mechanisms of Alzheimer disease is aiming at understanding how genes and their products modulate the pathogenesis of Alzheimer disease and finding therapeutic avenues.
Accumulation of amyloid β-peptide (Aβ) in extracellular plaques is a key feature of Alzheimer disease.Citation32 Aβ is derived by sequential proteolysis of the β-amyloid precursor protein. Synaptic dysfunction occurs early in the disease and there is a clear correlation between synaptic failure and dementia, and Aβ oligomers levels in the brain.Citation33-Citation35 PrPC was identified as a high affinity receptor for Aβ oligomers in an unbiased approach.Citation36 Importantly, PrPC/Aβ interactions were independently confirmed by different laboratories.Citation37-Citation40 The role of PrPC in Aβ-mediated toxicity in Alzheimer disease is currently a matter of contention. Some groups propose that PrPC mediates synaptic failure by Aβ oligomers,Citation36,Citation41-Citation49 while other laboratories challenge the function of PrPC in Aβ-mediated toxicity.Citation50-Citation53
Both charged region of PrPC (aa. 23–27 and 95–110) are essential for optimal interaction between PrPC and Aβ.Citation38,Citation39 Most importantly, these two regions are only present on PrPN1 and shed PrP. In several paradigms, these secreted PrPC molecules show promising activity against Aβ oligomers (). First, PrPN1 and shed PrP produced recombinantly or derived from cells display neuroprotective functions against Aβ mediated-toxicity.Citation11,Citation39,Citation54-Citation56 Second, both PrPN1 and to a lesser extent shed PrP, inhibit Aβ fibrillization, a critical step in the formation of toxic species of Aβ.Citation39,Citation56,Citation57 The absence of the second charged region on PrPN2 and the lower efficiency of shed PrP to inhibit Aβ fibrillization suggest that PrPN1 could represent the most efficient molecule interfering with Aβ fibrillization and toxicity. Third, PrPC1 does not bind Aβ oligomers which would also lead to a reduction of the neurodegeneration caused by Aβ. Again, it will be of great interest to use recombinant PrPN1 in vivo or to identify drugs capable of inducing PrPC α-cleavage in order to stop or reduce the development of Alzheimer disease.
PrPC N-terminal Domain/β-sheet Oligomers Interactions: Therapeutic Perspectives and Remaining Challenges
In recent years, unanticipated molecular cross talks between prion diseases and Alzheimer disease have emerged. PrPC may be a cell surface receptor for toxic soluble β-sheet oligomers, including Aβ oligomers, and may have an important role in synaptic dysfunction in Alzheimer disease. In animal models affected by both prion and Alzheimer disease, the pathogenesis of both diseases is clearly accelerated, suggesting a molecular connection between these two disorders.Citation58 The molecular mechanisms underlying these cross talks are incompletely understood and are debated in the literature. However, binding of β-sheet oligomers to PrPC N-terminal domain is undisputed, and the inhibition of Aβ oligomers toxicity by PrPN1 has been demonstrated in different laboratories.Citation11,Citation39,Citation54-Citation56
Two forms of PrPC N-terminal domain are naturally released by α- and β-cleavage, and there is some evidence that both α- and β-cleavage levels are increased in an animal model of Alzheimer disease.Citation59 We have shown that PrPC α-cleavage is increased in post-mortem human Alzheimer disease brains.Citation64 Thus, increased secretion of PrPC N-terminal domain may represent an endogenous compensatory mechanism aimed at interfering with Aβ-mediated toxicity.
In contrast to PrPN2, a neuroprotective activity of PrPN1 against β-sheet oligomers, including Aβ and PrPres oligomers and other insults was identified in different experimental settings,Citation11,Citation39,Citation54-Citation56,Citation60 and PrN1 is likely a promising start point molecule to develop therapeutic drugs. The protease responsible for PrPC α-cleavage is unknown and modulating the release of PrPN1 in the short-term represents a difficult challenge. In contrast, PrPN1 or peptides derived from PrPN1 may represent a unique opportunity to develop therapeutic molecules against prion and Alzheimer diseases. Such molecules would act as molecular traps and prevent β-sheet oligomers from binding to the cell surface and induce neuronal degeneration. Although PrPC N-terminal domain interacts with a broad range of ligands, unwanted side effects are unlikely because binding to PrPC N-terminal domain induces a molecular mechanism only when the N-terminal domain is physically connected to the C-terminal domain.Citation61
Several issues should be addressed before envisaging a specific therapeutic avenue. First, how does PrPN1 inhibit PrPres conversion and Aβ fibrillization? A simple mechanism could involve the formation of PrPN1/PrPres or PrPN1/Aβ complexes trapping PrPres and Aβ molecules and preventing further conversion and toxic effects. In agreement for this mechanism, PrPN1 association with Aβ causes a conformational change resulting in the formation of amorphous and insoluble aggregates that are not compatible with the amyloidogenic pathway leading to the assembly of Aβ oligomers and fibrils.Citation64 Another issue relates to the fate of PrPN1/PrPres or PrPN1/Aβ complexes. We have provided evidence for the presence of PrPN1/Aβ complexes in the guanidine-extractable fraction presumably representing insoluble amyloid plaques in post-mortem brain tissues of Alzheimer disease patients.Citation64 Interestingly, non-identified PrPC molecules were found in Alzheimer disease extracellular plaques.Citation62 These plaques were mainly recognized by antibodies targeting the N-terminal moiety of PrPC, and it is tempting to speculate that PrPC molecules found in these extracellular plaques of Alzheimer disease might be PrPN1. An important issue is the generation of a transgenic mouse constitutively expressing PrPN1. This mouse model is of paramount importance to test the therapeutic effect of PrPN1 in Alzheimer disease and prion diseases. However, PrPN1 is an unstructured polypeptide and targeting such polypeptide in the secretory pathway will be technically challenging.Citation63
In conclusion, recent data point to an emergent role of PrPC-derived natural proteolytic products as a source for new therapeutic drugs in prion and Alzheimer diseases. In particular, there is clear evidence that the fragment resulting from PrPC α-cleavage, PrPN1 has protective activity in vivo and has strong therapeutic potential. We predict that the development of a transgenic mouse expressing PrPN1 will be crucial importance to make a Go/No-Go decision in PrPN1-based drug development.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
This research was supported in part by a grant from the Fonds de Recherche en Santé, Québec and the Canadian Institutes for Health Research (MOP-89881) to X.R. We thank Julie Motard and Guillaume Tremblay for critical reading of the manuscript.
Related Research Data
References
- Prusiner SB, Scott MR, DeArmond SJ, Cohen FE. Prion protein biology. Cell 1998; 93:337 - 48; http://dx.doi.org/10.1016/S0092-8674(00)81163-0; PMID: 9590169
- Marijanovic Z, Caputo A, Campana V, Zurzolo C. Identification of an intracellular site of prion conversion. PLoS Pathog 2009; 5:e1000426; http://dx.doi.org/10.1371/journal.ppat.1000426; PMID: 19424437
- Goold R, Rabbanian S, Sutton L, Andre R, Arora P, Moonga J, Clarke AR, Schiavo G, Jat P, Collinge J, et al. Rapid cell-surface prion protein conversion revealed using a novel cell system. Nat Commun 2011; 2:281; http://dx.doi.org/10.1038/ncomms1282; PMID: 21505437
- Mallucci G, Dickinson A, Linehan J, Klöhn PC, Brandner S, Collinge J. Depleting neuronal PrP in prion infection prevents disease and reverses spongiosis. Science 2003; 302:871 - 4; http://dx.doi.org/10.1126/science.1090187; PMID: 14593181
- Zahn R, Liu A, Lührs T, Riek R, von Schroetter C, López García F, Billeter M, Calzolai L, Wider G, Wüthrich K. NMR solution structure of the human prion protein. Proc Natl Acad Sci U S A 2000; 97:145 - 50; http://dx.doi.org/10.1073/pnas.97.1.145; PMID: 10618385
- Yuan J, Zhan YA, Abskharon R, Xiao X, Martinez MC, Zhou X, Kneale G, Mikol J, Lehmann S, Surewicz WK, et al. Recombinant human prion protein inhibits prion propagation in vitro. Sci Rep 2013; 3:2911; http://dx.doi.org/10.1038/srep02911; PMID: 24105336
- Turnbaugh JA, Unterberger U, Saá P, Massignan T, Fluharty BR, Bowman FP, Miller MB, Supattapone S, Biasini E, Harris DA. The N-terminal, polybasic region of PrP(C) dictates the efficiency of prion propagation by binding to PrP(Sc). J Neurosci 2012; 32:8817 - 30; http://dx.doi.org/10.1523/JNEUROSCI.1103-12.2012; PMID: 22745483
- Büeler H, Aguzzi A, Sailer A, Greiner RA, Autenried P, Aguet M, Weissmann C. Mice devoid of PrP are resistant to scrapie. Cell 1993; 73:1339 - 47; http://dx.doi.org/10.1016/0092-8674(93)90360-3; PMID: 8100741
- Brandner S, Isenmann S, Raeber A, Fischer M, Sailer A, Kobayashi Y, Marino S, Weissmann C, Aguzzi A. Normal host prion protein necessary for scrapie-induced neurotoxicity. Nature 1996; 379:339 - 43; http://dx.doi.org/10.1038/379339a0; PMID: 8552188
- Rambold AS, Müller V, Ron U, Ben-Tal N, Winklhofer KF, Tatzelt J. Stress-protective signalling of prion protein is corrupted by scrapie prions. EMBO J 2008; 27:1974 - 84; http://dx.doi.org/10.1038/emboj.2008.122; PMID: 18566584
- Resenberger UK, Harmeier A, Woerner AC, Goodman JL, Müller V, Krishnan R, Vabulas RM, Kretzschmar HA, Lindquist S, Hartl FU, et al. The cellular prion protein mediates neurotoxic signalling of β-sheet-rich conformers independent of prion replication. EMBO J 2011; 30:2057 - 70; http://dx.doi.org/10.1038/emboj.2011.86; PMID: 21441896
- Mangé A, Béranger F, Peoc’h K, Onodera T, Frobert Y, Lehmann S. Alpha- and beta- cleavages of the amino-terminus of the cellular prion protein. Biol Cell 2004; 96:125 - 32; http://dx.doi.org/10.1016/j.biolcel.2003.11.007; PMID: 15050367
- Chen SG, Teplow DB, Parchi P, Teller JK, Gambetti P, Autilio-Gambetti L. Truncated forms of the human prion protein in normal brain and in prion diseases. J Biol Chem 1995; 270:19173 - 80; http://dx.doi.org/10.1074/jbc.270.32.19173; PMID: 7642585
- Altmeppen HC, Prox J, Puig B, Dohler F, Falker C, Krasemann S, Glatzel M. Roles of endoproteolytic α-cleavage and shedding of the prion protein in neurodegeneration. FEBS J 2013; 280:4338 - 47; http://dx.doi.org/10.1111/febs.12196; PMID: 23413979
- Stahl N, Baldwin MA, Burlingame AL, Prusiner SB. Identification of glycoinositol phospholipid linked and truncated forms of the scrapie prion protein. Biochemistry 1990; 29:8879 - 84; http://dx.doi.org/10.1021/bi00490a001; PMID: 1980209
- Taylor DR, Parkin ET, Cocklin SL, Ault JR, Ashcroft AE, Turner AJ, Hooper NM. Role of ADAMs in the ectodomain shedding and conformational conversion of the prion protein. J Biol Chem 2009; 284:22590 - 600; http://dx.doi.org/10.1074/jbc.M109.032599; PMID: 19564338
- Tagliavini F, Prelli F, Porro M, Salmona M, Bugiani O, Frangione B. A soluble form of prion protein in human cerebrospinal fluid: implications for prion-related encephalopathies. Biochem Biophys Res Commun 1992; 184:1398 - 404; http://dx.doi.org/10.1016/S0006-291X(05)80038-5; PMID: 1375461
- Parizek P, Roeckl C, Weber J, Flechsig E, Aguzzi A, Raeber AJ. Similar turnover and shedding of the cellular prion protein in primary lymphoid and neuronal cells. J Biol Chem 2001; 276:44627 - 32; http://dx.doi.org/10.1074/jbc.M107458200; PMID: 11571302
- Parkin ET, Watt NT, Turner AJ, Hooper NM. Dual mechanisms for shedding of the cellular prion protein. J Biol Chem 2004; 279:11170 - 8; http://dx.doi.org/10.1074/jbc.M312105200; PMID: 14711812
- Watt NT, Hooper NM. Reactive oxygen species (ROS)-mediated beta-cleavage of the prion protein in the mechanism of the cellular response to oxidative stress. Biochem Soc Trans 2005; 33:1123 - 5; http://dx.doi.org/10.1042/BST20051123; PMID: 16246061
- Harris DA, Huber MT, van Dijken P, Shyng SL, Chait BT, Wang R. Processing of a cellular prion protein: identification of N- and C-terminal cleavage sites. Biochemistry 1993; 32:1009 - 16; http://dx.doi.org/10.1021/bi00055a003; PMID: 8093841
- Chesebro B, Trifilo M, Race R, Meade-White K, Teng C, LaCasse R, Raymond L, Favara C, Baron G, Priola S, et al. Anchorless prion protein results in infectious amyloid disease without clinical scrapie. Science 2005; 308:1435 - 9; http://dx.doi.org/10.1126/science.1110837; PMID: 15933194
- Klingeborn M, Race B, Meade-White KD, Rosenke R, Striebel JF, Chesebro B. Crucial role for prion protein membrane anchoring in the neuroinvasion and neural spread of prion infection. J Virol 2011; 85:1484 - 94; http://dx.doi.org/10.1128/JVI.02167-10; PMID: 21123371
- McNally KL, Ward AE, Priola SA. Cells expressing anchorless prion protein are resistant to scrapie infection. J Virol 2009; 83:4469 - 75; http://dx.doi.org/10.1128/JVI.02412-08; PMID: 19225008
- Meier P, Genoud N, Prinz M, Maissen M, Rülicke T, Zurbriggen A, Raeber AJ, Aguzzi A. Soluble dimeric prion protein binds PrP(Sc) in vivo and antagonizes prion disease. Cell 2003; 113:49 - 60; http://dx.doi.org/10.1016/S0092-8674(03)00201-0; PMID: 12679034
- Westergard L, Turnbaugh JA, Harris DA. A naturally occurring C-terminal fragment of the prion protein (PrP) delays disease and acts as a dominant-negative inhibitor of PrPSc formation. J Biol Chem 2011; 286:44234 - 42; http://dx.doi.org/10.1074/jbc.M111.286195; PMID: 22025612
- Yadavalli R, Guttmann RP, Seward T, Centers AP, Williamson RA, Telling GC. Calpain-dependent endoproteolytic cleavage of PrPSc modulates scrapie prion propagation. J Biol Chem 2004; 279:21948 - 56; http://dx.doi.org/10.1074/jbc.M400793200; PMID: 15026410
- Pietri M, Dakowski C, Hannaoui S, Alleaume-Butaux A, Hernandez-Rapp J, Ragagnin A, Mouillet-Richard S, Haik S, Bailly Y, Peyrin JM, et al. PDK1 decreases TACE-mediated α-secretase activity and promotes disease progression in prion and Alzheimer’s diseases. Nat Med 2013; 19:1124 - 31; http://dx.doi.org/10.1038/nm.3302; PMID: 23955714
- Aguzzi A, Baumann F, Bremer J. The prion’s elusive reason for being. Annu Rev Neurosci 2008; 31:439 - 77; http://dx.doi.org/10.1146/annurev.neuro.31.060407.125620; PMID: 18558863
- Chesebro B, Race B, Meade-White K, Lacasse R, Race R, Klingeborn M, Striebel J, Dorward D, McGovern G, Jeffrey M. Fatal transmissible amyloid encephalopathy: a new type of prion disease associated with lack of prion protein membrane anchoring. PLoS Pathog 2010; 6:e1000800; http://dx.doi.org/10.1371/journal.ppat.1000800; PMID: 20221436
- Alzheimer's disease international world alzheimer report 2013. 2013:1-92.
- Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 1991; 82:239 - 59; http://dx.doi.org/10.1007/BF00308809; PMID: 1759558
- Lue LF, Kuo YM, Roher AE, Brachova L, Shen Y, Sue L, Beach T, Kurth JH, Rydel RE, Rogers J. Soluble amyloid beta peptide concentration as a predictor of synaptic change in Alzheimer’s disease. Am J Pathol 1999; 155:853 - 62; http://dx.doi.org/10.1016/S0002-9440(10)65184-X; PMID: 10487842
- McLean CA, Cherny RA, Fraser FW, Fuller SJ, Smith MJ, Beyreuther K, Bush AI, Masters CL. Soluble pool of Abeta amyloid as a determinant of severity of neurodegeneration in Alzheimer’s disease. Ann Neurol 1999; 46:860 - 6; http://dx.doi.org/10.1002/1531-8249(199912)46:6<860::AID-ANA8>3.0.CO;2-M; PMID: 10589538
- Selkoe DJ. Alzheimer’s disease is a synaptic failure. Science 2002; 298:789 - 91; http://dx.doi.org/10.1126/science.1074069; PMID: 12399581
- Laurén J, Gimbel DA, Nygaard HB, Gilbert JW, Strittmatter SM. Cellular prion protein mediates impairment of synaptic plasticity by amyloid-beta oligomers. Nature 2009; 457:1128 - 32; http://dx.doi.org/10.1038/nature07761; PMID: 19242475
- Bate C, Williams A. Amyloid-β-induced synapse damage is mediated via cross-linkage of cellular prion proteins. J Biol Chem 2011; 286:37955 - 63; http://dx.doi.org/10.1074/jbc.M111.248724; PMID: 21900234
- Chen S, Yadav SP, Surewicz WK. Interaction between human prion protein and amyloid-beta (Abeta) oligomers: role OF N-terminal residues. J Biol Chem 2010; 285:26377 - 83; http://dx.doi.org/10.1074/jbc.M110.145516; PMID: 20576610
- Fluharty BR, Biasini E, Stravalaci M, Sclip A, Diomede L, Balducci C, La Vitola P, Messa M, Colombo L, Forloni G, et al. An N-terminal fragment of the prion protein binds to amyloid-β oligomers and inhibits their neurotoxicity in vivo. J Biol Chem 2013; 288:7857 - 66; http://dx.doi.org/10.1074/jbc.M112.423954; PMID: 23362282
- Zou WQ, Xiao X, Yuan J, Puoti G, Fujioka H, Wang X, Richardson S, Zhou X, Zou R, Li S, et al. Amyloid-beta42 interacts mainly with insoluble prion protein in the Alzheimer brain. J Biol Chem 2011; 286:15095 - 105; http://dx.doi.org/10.1074/jbc.M110.199356; PMID: 21393248
- Alier K, Ma L, Yang J, Westaway D, Jhamandas JH. Aβ inhibition of ionic conductance in mouse basal forebrain neurons is dependent upon the cellular prion protein PrPC. J Neurosci 2011; 31:16292 - 7; http://dx.doi.org/10.1523/JNEUROSCI.4367-11.2011; PMID: 22072680
- You H, Tsutsui S, Hameed S, Kannanayakal TJ, Chen L, Xia P, Engbers JD, Lipton SA, Stys PK, Zamponi GW. Aβ neurotoxicity depends on interactions between copper ions, prion protein, and N-methyl-D-aspartate receptors. Proc Natl Acad Sci U S A 2012; 109:1737 - 42; http://dx.doi.org/10.1073/pnas.1110789109; PMID: 22307640
- Barry AE, Klyubin I, Mc Donald JM, Mably AJ, Farrell MA, Scott M, Walsh DM, Rowan MJ. Alzheimer’s disease brain-derived amyloid-β-mediated inhibition of LTP in vivo is prevented by immunotargeting cellular prion protein. J Neurosci 2011; 31:7259 - 63; http://dx.doi.org/10.1523/JNEUROSCI.6500-10.2011; PMID: 21593310
- Freir DB, Nicoll AJ, Klyubin I, Panico S, Mc Donald JM, Risse E, Asante EA, Farrow MA, Sessions RB, Saibil HR, et al. Interaction between prion protein and toxic amyloid β assemblies can be therapeutically targeted at multiple sites. Nat Commun 2011; 2:336; http://dx.doi.org/10.1038/ncomms1341; PMID: 21654636
- Gimbel DA, Nygaard HB, Coffey EE, Gunther EC, Laurén J, Gimbel ZA, Strittmatter SM. Memory impairment in transgenic Alzheimer mice requires cellular prion protein. J Neurosci 2010; 30:6367 - 74; http://dx.doi.org/10.1523/JNEUROSCI.0395-10.2010; PMID: 20445063
- Kudo W, Lee HP, Zou WQ, Wang X, Perry G, Zhu X, Smith MA, Petersen RB, Lee HG. Cellular prion protein is essential for oligomeric amyloid-β-induced neuronal cell death. Hum Mol Genet 2012; 21:1138 - 44; http://dx.doi.org/10.1093/hmg/ddr542; PMID: 22100763
- Ostapchenko VG, Beraldo FH, Mohammad AH, Xie YF, Hirata PH, Magalhaes AC, Lamour G, Li H, Maciejewski A, Belrose JC, et al. The prion protein ligand, stress-inducible phosphoprotein 1, regulates amyloid-β oligomer toxicity. J Neurosci 2013; 33:16552 - 64; http://dx.doi.org/10.1523/JNEUROSCI.3214-13.2013; PMID: 24133259
- Um JW, Kaufman AC, Kostylev M, Heiss JK, Stagi M, Takahashi H, Kerrisk ME, Vortmeyer A, Wisniewski T, Koleske AJ, et al. Metabotropic glutamate receptor 5 is a coreceptor for Alzheimer aβ oligomer bound to cellular prion protein. Neuron 2013; 79:887 - 902; http://dx.doi.org/10.1016/j.neuron.2013.06.036; PMID: 24012003
- Um JW, Nygaard HB, Heiss JK, Kostylev MA, Stagi M, Vortmeyer A, Wisniewski T, Gunther EC, Strittmatter SM. Alzheimer amyloid-β oligomer bound to postsynaptic prion protein activates Fyn to impair neurons. Nat Neurosci 2012; 15:1227 - 35; http://dx.doi.org/10.1038/nn.3178; PMID: 22820466
- Balducci C, Beeg M, Stravalaci M, Bastone A, Sclip A, Biasini E, Tapella L, Colombo L, Manzoni C, Borsello T, et al. Synthetic amyloid-beta oligomers impair long-term memory independently of cellular prion protein. Proc Natl Acad Sci U S A 2010; 107:2295 - 300; http://dx.doi.org/10.1073/pnas.0911829107; PMID: 20133875
- Calella AM, Farinelli M, Nuvolone M, Mirante O, Moos R, Falsig J, Mansuy IM, Aguzzi A. Prion protein and Abeta-related synaptic toxicity impairment. EMBO Mol Med 2010; 2:306 - 14; http://dx.doi.org/10.1002/emmm.201000082; PMID: 20665634
- Cissé M, Sanchez PE, Kim DH, Ho K, Yu GQ, Mucke L. Ablation of cellular prion protein does not ameliorate abnormal neural network activity or cognitive dysfunction in the J20 line of human amyloid precursor protein transgenic mice. J Neurosci 2011; 31:10427 - 31; http://dx.doi.org/10.1523/JNEUROSCI.1459-11.2011; PMID: 21775587
- Kessels HW, Nguyen LN, Nabavi S, Malinow R. The prion protein as a receptor for amyloid-beta. Nature 2010; 466:E3 - 4, discussion E4-5; http://dx.doi.org/10.1038/nature09217; PMID: 20703260
- Béland M, Motard J, Barbarin A, Roucou X. PrP(C) homodimerization stimulates the production of PrPC cleaved fragments PrPN1 and PrPC1. J Neurosci 2012; 32:13255 - 63; http://dx.doi.org/10.1523/JNEUROSCI.2236-12.2012; PMID: 22993441
- Guillot-Sestier MV, Sunyach C, Ferreira ST, Marzolo MP, Bauer C, Thevenet A, Checler F. α-Secretase-derived fragment of cellular prion, N1, protects against monomeric and oligomeric amyloid β (Aβ)-associated cell death. J Biol Chem 2012; 287:5021 - 32; http://dx.doi.org/10.1074/jbc.M111.323626; PMID: 22184125
- Nieznanski K, Choi JK, Chen S, Surewicz K, Surewicz WK. Soluble prion protein inhibits amyloid-β (Aβ) fibrillization and toxicity. J Biol Chem 2012; 287:33104 - 8; http://dx.doi.org/10.1074/jbc.C112.400614; PMID: 22915585
- Younan ND, Sarell CJ, Davies P, Brown DR, Viles JH. The cellular prion protein traps Alzheimer’s Aβ in an oligomeric form and disassembles amyloid fibers. FASEB J 2013; 27:1847 - 58; http://dx.doi.org/10.1096/fj.12-222588; PMID: 23335053
- Morales R, Estrada LD, Diaz-Espinoza R, Morales-Scheihing D, Jara MC, Castilla J, Soto C. Molecular cross talk between misfolded proteins in animal models of Alzheimer’s and prion diseases. J Neurosci 2010; 30:4528 - 35; http://dx.doi.org/10.1523/JNEUROSCI.5924-09.2010; PMID: 20357103
- Ostapchenko VG, Beraldo FH, Guimarães AL, Mishra S, Guzman M, Fan J, Martins VR, Prado VF, Prado MA. Increased prion protein processing and expression of metabotropic glutamate receptor 1 in a mouse model of Alzheimer’s disease. J Neurochem 2013; 127:415 - 25; http://dx.doi.org/10.1111/jnc.12296; PMID: 23651058
- Guillot-Sestier MV, Sunyach C, Druon C, Scarzello S, Checler F. The alpha-secretase-derived N-terminal product of cellular prion, N1, displays neuroprotective function in vitro and in vivo. J Biol Chem 2009; 284:35973 - 86; http://dx.doi.org/10.1074/jbc.M109.051086; PMID: 19850936
- Béland M, Roucou X. The prion protein unstructured N-terminal region is a broad-spectrum molecular sensor with diverse and contrasting potential functions. J Neurochem 2012; 120:853 - 68; PMID: 22145935
- Privat N, Laffont-Proust I, Faucheux BA, Sazdovitch V, Frobert Y, Laplanche JL, Grassi J, Hauw JJ, Haïk S. Human prion diseases: from antibody screening to a standardized fast immunodiagnosis using automation. Mod Pathol 2008; 21:140 - 9; PMID: 18084251
- Pfeiffer NV, Dirndorfer D, Lang S, Resenberger UK, Restelli LM, Hemion C, Miesbauer M, Frank S, Neutzner A, Zimmermann R, et al. Structural features within the nascent chain regulate alternative targeting of secretory proteins to mitochondria. EMBO J 2013; 32:1036 - 51; http://dx.doi.org/10.1038/emboj.2013.46; PMID: 23481258
- Béland M, Bédard M, Tremblay G, Lavigne P, Roucou X. Aβ induces its own prion protein N-terminal fragment (PrPN1)-mediated neutralization in amorphous aggregates. Neurobiol Aging 2014; http://dx.doi.org/10.1016/j.neurobiolaging.2014.02.001