Abstract
The capacity to polymerize into amyloid fibrils is common to many proteins. While some proteins naturally form these fibrils to serve functional roles, amyloid is usually associated with pathogenic processes in which specific proteins aberrantly aggregate within cells or tissues. Though the contribution of amyloid fibrils to actual disease pathogenesis is not always clear, one possibility is that the titration of essential proteins from solution into aggregates contributes to the cellular degeneration common to many amyloid diseases. Using mammalian and yeast model systems, we recently showed that the common biophysical properties of amyloid aggregates—including strong resistance to dissolution—enable stringent purification and identification of both amyloid-forming and amyloid-associated proteins directly from cells. Strikingly, many proteins that were previously implicated in formation or clearance of intracellular aggregates, including several stress granule components, were found to co-aggregate with amyloid formed by a polyglutamine-expanded huntingtin fragment. This direct evaluation of proteins within aggregates can help identify new amyloid-forming proteins, as well as proteins that can indirectly contribute to disease mechanisms.
Introduction
Amyloid is a filamentous homo-polymeric protein aggregate with repetitive structural order, much like a one-dimensional crystal.Citation1 Its structure is based on the stacking of polypeptide chains that lie orthogonal to the long fibril axis, forming extended β sheets that run the length of the aggregate (cross-β structure). The extensive hydrogen bonding network and the face-to-face alignment of β sheets (steric zippers) result in robust structures that can exhibit a very high degree of resistance to detergents and proteases.Citation2 Moreover, individual fibrils can associate laterally to form large superstructures with low surface-to-volume ratios,Citation3 thus further protecting the constitutive proteins from denaturants.
Several natural examples exist in which the structural properties of amyloid are harnessed for specific functions.Citation4 The quintessential example of a so-called beneficial amyloid is the bacterial protein CsgA, which assembles into very robust extracellular fibers that are a component of biofilms.Citation5 Despite numerous reports of beneficial amyloid fibrils, amyloid continues to be more associated with pathogenic processes. This is especially due to the huge societal cost of amyloid diseases, which increase sharply in prevalence in older populations. Alzheimer disease, which features the accumulation of β-amyloid peptides in the brain, is the most notorious of the amyloid disorders, though dozens of other diseases are also linked to the accumulation of specific proteins into similar aggregates.Citation6
Conversion of a protein into pathological amyloid is generally considered an aberrant stochastic event, but once established, fibrils grow by template-driven addition at their ends, with high specificity for protein of identical amino-acid sequence. However, under rare circumstances, two proteins may co-polymerize within one fibril,Citation7 usually in cases where the sequences of two proteins are closely related. Similarly, some amyloid fibrils can “cross-seed,” i.e. potentiate the amyloid conversion of a different protein.Citation8 Consequentially, both co-polymerization and cross-seeding can result in protein inclusions that contain more than one aggregated species; in such cases, recognizing the protein responsible for the primary aggregation event may be difficult. Also, the recruitment and mislocalization of secondary proteins to aggregates may have implications in disease mechanisms ().
Figure 1. Cartoon representation of an isogenic pair of yeast cells. The cell on the right harbors an amyloid aggregate. TAPI (technique for amyloid purification and identification) couples a novel purification scheme with tandem mass spectrometry to identify proteins that form amyloid or are tightly associated with amyloid aggregates.
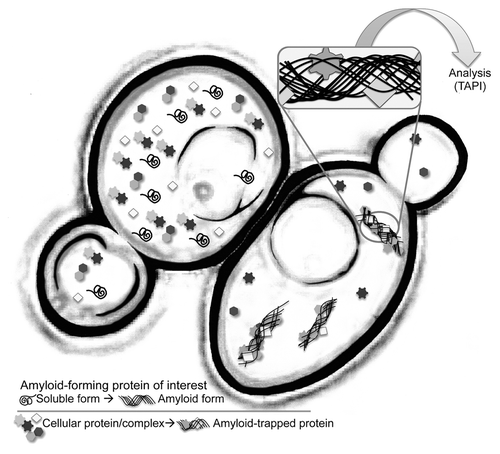
Identifying new amyloid-forming and amyloid-associated proteins is of fundamental interest. However, because amyloid is a low-energy conformation that can be adopted by many different polypeptides of very different amino acid composition, predicting which proteins will form amyloid can be challenging. No particular protein sequence precisely determines if a protein will form amyloid in its native environment (excluding proteins that form amyloid as part of their normal function), so it can be challenging to use bioinformatic approaches to predict amyloid-forming proteins, especially considering the many cellular and systemic conditions that ultimately influence a protein’s fate. Even very good bioinformatic strategies can have weaknesses, such as being limited to glutamine/asparagine-rich proteins.Citation9 We recently developed a new strategy that directly assays for the presence of aggregates that possess the archetypical properties of amyloid. The same biophysical properties that are problematic from a protein-quality control perspective—such as large size, resistance to dissolution and ability to self-propagate—can be exploited for the isolation and identification of proteins within (or tightly attached to) amyloid aggregates. Here we overview a strategy we call TAPI (technique for amyloid protein identificationCitation10) and discuss its advantages and limitations for identifying amyloid-forming and amyloid-associating proteins.
Non-Targeted Identification of Yeast Prions Using TAPI
The yeast Saccharomyces cerevisiae naturally encodes at least 7 different intracellular proteins that can adopt self-propagating amyloid forms.Citation11 These proteins are collectively known as prions (infectious proteins) because their amyloid conformations can be transmitted to other cells during cellular division or mating. The three best studied examples are: the [PSI+] prion formed by the Sup35 protein, [URE3] formed by Ure2, and [RNQ+] formed by Rnq1. These prions have proven to be an excellent model for both amyloid and prion diseasesCitation11 since yeast cells can often harbor them with little physiological detriment.Citation12 Importantly, yeast prion aggregates, despite being formed by different proteins, share certain biophysical properties typical of amyloid. For example, the protein-protein interactions within Sup35 prion aggregates have unusual resistance to protease digestion, urea, and sodium dodecyl sulfate (SDS).Citation13-Citation16 While most proteins are denatured by 2% SDS, especially at higher temperatures, the integrity of Sup35 amyloid aggregates resist prolonged SDS treatment, even under conditions where most yeast protein complexes are destroyed.Citation10,Citation16
With TAPI, we exploited this SDS-resistance and also employed a novel method for separating the high molecular weight amyloid species from other protein complexes.Citation10 Since low abundance and extreme heterogeneity of yeast prion amyloid precludes its separation by standard chromatography methods (our unpublished observation), we separated and enriched aggregates by trapping them at the top of gradient acrylamide gels using standard SDS-PAGE, followed by elution under denaturing conditions.Citation10 During electrophoresis, the migration of SDS-coated aggregates is retarded, while solubilized proteins will migrate further into the gel. After electrophoresis, the region containing the trapped high molecular weight SDS-resistant aggregates is excised and subjected to prolonged incubation with SDS at near-boiling temperatures. The eluted proteins are then concentrated, purified from salts and SDS, digested with specific proteases (Trypsin), and analyzed by tandem mass-spectrometry.
By comparing mass spectrometry results with negative controls (e.g., isogenic strains without amyloid aggregates), we successfully identified the three most-studied yeast prions from both laboratory and wild strains, as well as aggregates formed by huntingtin exon 1 fragment (discussed below). The amyloid-forming proteins scored highly (i.e. many identifiable peptides by mass spectrometry) among all identified proteins, likely due to the abundance of peptides generated from amyloid polymer. An important advantage of this strategy is the ability to analyze amyloid material from a variety of sources with high stringency and specificity (). Only the largest, most resistant complexes survive the isolation procedure, thus greatly reducing the number of irrelevant hits. Moreover, this approach is non-targeted, requiring no specific antibodies. Neither does it require subjective comparisons between stained gels, accompanied by individual gel-band dissection and analysis. A limitation of TAPI is its bias for aggregates with specific properties: aggregates should be high molecular weight and resistant (at least partially) to detergent treatment. For example, amyloid oligomers may escape detection if they fall below a certain size threshold and migrate further into the gel. Likewise, not all amyloid fibrils exhibit the same degree of resistance to chaotropic agents, thus SDS-sensitive aggregates will also escape identification.
Application of TAPI for New Amyloid Discovery
The discovery of new fungal prions was our initial impetus for developing a non-targeted method for identification of amyloid-forming proteins. Our methodology is well suited to expand the study of prion/amyloid-based epigenetics beyond laboratory strains of S. cerevisiae. Prions are unique epigenetic phenomena because the activity levels of prion proteins switch with their physical state; two genetically-identical cells can differ phenotypically depending on the status of any given prion protein. With the exception of mammalian prion protein and the [Het-s] prion of Podospora anserina,Citation17 the ubiquity of prions in other organisms remains unknown. Despite some limitations (see next paragraph), TAPI offers an unbiased amyloid identification strategy for less tractable organisms. Several intriguing non-Mendelian phenomena have been described in fungi, including phenotypic switching and dimorphism.Citation18,Citation19 It would be attractive to test whether amyloid-based prion mechanisms underlie some of these observed phenotypes.
In the case of known amyloid-forming proteins, the effectiveness of TAPI can be verified by standard immuno-blotting of trapped protein species using specific antibodies prior to mass spectrometry analysis. However, in the pursuit of discovering novel amyloid proteins, specific antibodies are not an option, and amyloid enrichment conditions cannot be easily optimized. Therefore, TAPI may deliver many potential candidates. This is partially due to the inherent complexity of mass spectrometry, but proper negative controls will help eliminate contaminant species that are frequently present in SDS-resistant fractions. Negative controls might include cells or organisms treated with prion-eliminating agents, or simply isogenic organisms with distinct phenotypes. By repeating control samples several times, a reliable list of contaminant proteins can be generated, which enables stringent discrimination when analyzing potential amyloid-containing samples. However, amyloid-forming proteins could escape identification if they are only in a minority of the sample population—due to inherent instability of some amyloid-based prions—or for reasons previously discussed. Once strong candidate proteins are identified, established secondary assays are necessary to determine if the proteins are indeed forming amyloid in vivo. Such assays generally involve monitoring a protein for a transition into a high molecular-weight species with distinct biophysical properties.Citation20 Examples include sedimentation analysis, semi-denaturing detergent agarose electrophoreses (SDD-AGE), fluorescence microscopy localization, and filter-retardation assays. The capacity of a candidate protein to form amyloid can also be determined in vitro with recombinant protein.
Wild cerevisiae strains may possess uncharacterized amyloid-based prions that are responsible for inherited phenotypic variations.Citation21 Using TAPI, we evaluated a portion (about 10 strains; publishedCitation10 and unpublished results) from a large collection of wild yeast strains that was reported to likely contain unknown prionsCitation21 (based on the observation that some strains developed new phenotypes following exposure to guanidine hydrochloride, a compound that inhibits the passage of prion amyloid to progeny cells). We found the published phenotypic differences to be very subtle or below detection limits. Furthermore, of the stains we analyzed by TAPI, we did not identify promising candidates that were unique to those strains while absent from control strains. We cannot exclude the possibility of prions existing in these tested strains because they could possess non-classical properties (SDS-sensitivity, non-amyloid-based, hyper-instability), and thus escape detection. Nevertheless we have identified ~10–15 proteins that are consistently present in SDS-resistant fractions prepared from several unrelated yeast strainsCitation10 (and our unpublished results). It would be interesting to know if these proteins are forming intracellular amyloid, and if the formation of stable structural assemblies is important for their biological functions. For example, the protein Ygr130c was identified from several strains. This protein contains extended intrinsically disordered regions and is part of the eisosome,Citation22 a very large hetero-complex at the plasma membrane that facilitates endocytosis.Citation23 Conceivably, Ygr130c—and other proteins that form macromolecular structures—could employ amyloid-like interactions for self-assembly or scaffolding.
There has been controversy about whether or not prions may provide a utility for yeast cells.Citation24 It was proposed that the presence of prions, being mildly toxic under normal growth conditions, can provide benefits to cells during various stresses. In particular, rapid and reversible prion conversion might serve as an evolutionary mechanism that promotes short-term survival under fluctuating environmental conditions.Citation25,Citation26 Prion induction under stress would be promoted by increased protein misfolding and decreased protein quality-control efficiency, or by increased expression of chaperones that facilitate prion conversion.Citation27 Utilizing this mechanism, cells could temporarily alter their functional proteome without underlying genomic changes. In support of this hypothesis, it was reported that severe stress conditions increased the rate of de novo [PSI+] generation,Citation25 although we could not independently confirm this observation.Citation28 If this hypothesis is true, prions should arise and be stabilized under stress conditions. TAPI offers an opportunity to test whether yeast strains subjected to selective pressures could generate specific SDS-resistant protein profiles. Established secondary assays could then be used for the validation of prion/amyloid-forming candidates.
Potential for Structural Characterization of Natural Amyloid Fibrils
One surprising observation during the development of the TAPI approach was the large amount of pure Sup35 amyloid that could be isolated from [PSI+] cells. After elution under denaturing conditions, the amyloid preparation from [PSI+] cells yielded almost exclusively Sup35 protein.10We calculated that milligram quantities of amyloid were extractable from a few liters of culture. This material could be used for direct structural characterization, eliminating the need to produce amyloid from recombinant protein, since in vitro preparation of fibrils tends to give morphologically heterogeneous mixtures with unknown resemblance to in vivo species. Thus far, solid-state NMR has proven an exceptional method for studying the structure of recombinant isotope-labeled yeast prion amyloid.Citation29 Similar regimens used for isotope labeling in bacterial culture can be applied to yeast cells, followed by the TAPI isolation protocol to produce ex vivo amyloid samples for solid-state NMR studies.
Isolation and Identification of Proteins Strongly Associated with Human Amyloids
In addition to new amyloid identification, perhaps the greatest potential of TAPI lies in discovery or confirmation of amyloid-associated proteins. Our preliminary results suggest we can identify proteins that are uniquely recruited to specific types of protein aggregates. We first observed this with Sis1—a protein known to interact with the Rnq1 prion proteinCitation30,Citation31—when we identified it with Rnq1 from [PIN+] cells using TAPI.Citation10 While the interaction between Sis1 and Rnq1 may be particularly strong, other amyloid-interacting proteins may have weaker affinities. Also, co-polymerized/co-aggregated species might be present in lower concentrations. Therefore, optimization must be considered when working with a model amyloid. Stringency of the detergent treatment (detergent type and concentration) during TAPI may be adjusted to provide some flexibility for less tightly-associated proteins.
Co-aggregating species were identified by TAPI from yeast cells expressing HttQ103-GFP, a model protein that contains exon 1 of human huntingtin protein with an expanded polyglutamine tract. This protein readily forms amyloid during both yeast and mammalian expression with properties that resemble yeast prion aggregates, particularly SDS resistance. We identified a specific set of proteins present in the SDS-resistant fractions from cells expressing HttQ103-GFP, but not in control cells.Citation10 Interestingly, we found the stress-granule protein Pub1 was tightly associated with polyglutamine aggregates. Further analysis by fluorescence microscopy found that some yeast stress granule proteins were specifically recruited to polyglutamine aggregates, while others were not. Aberrant recruitment of certain stress granule components to aggregates may disturb RNA homeostasis and contribute to the pathological cellular mechanisms that underlie Huntington disease.Citation32 As a continuation of this work, we recently completed a comprehensive comparison of proteins associated specifically with HttQ74-GFP amyloid isolated from mammalian PC12 cells (undifferentiated cells of neuronal origin). We found four cellular pathways to be overrepresented in the results (publishedCitation10 and unpublished results): RNA processing, intracellular trafficking, mitochondrial and protein quality control. Intriguingly, specific amyotrophic lateral sclerosis (ALS)-associated proteins (FUS and TDP-43) were also found by TAPI to co-aggregate with polyglutamine. Overall, we observed a reduced total number of hits compared with other published studies,Citation32,Citation33 while also identifying several proteins that were not previously known to interact with polyglutamine aggregates, including other ALS-linked proteins (unpublished data).
We are confident about the quality and stringency of initial TAPI data sets. The method was developed to achieve mass spectrometry results without rigorous differential band staining or band excision. Further, extensive detergent treatment and filtration of high molecular weight species ensures survival of only the strongest interactors. As a result, we achieve a tight group of candidates that simplifies subsequent data analysis by reducing the burden of validating several hundred hits. We suggest that some of the identified co-aggregated proteins, observed in mammalian cells, may contribute to Huntington disease pathology, similar to the observed recruitment of glutamine-rich transcriptional factors to polyglutamine aggregates.Citation34,Citation35
A further direction for these studies is to analyze different types of amyloid aggregates, either extracellular or intracellular, from various cell and tissue sources. Comparative proteomic profiles generated by TAPI can show which proteins are commonly recruited to aggregates across several disease models, and which are unique to specific types of aggregates. This approach can identify cellular machinery that is typically involved in amyloid formation or clearance. Validation of interesting candidates may help elucidate pathological mechanisms, or conversely, identify specific factors involved in elimination of aggregates. Ultimately, the unbiased nature of TAPI lends itself to studying many disorders and phenomena associated with protein aggregation and amyloid formation.
| Abbreviations: | ||
| TAPI | = | technique for amyloid purification and identification |
| SDS | = | sodium dodecyl sulfate |
| PAGE | = | polyacrylamide gel electrophoresis |
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank R Wickner, H Edskes, A Kelly, B Burnett, and N Pripuzova for helpful discussion.
References
- Greenwald J, Riek R. Biology of amyloid: structure, function, and regulation. Structure 2010; 18:1244 - 60; http://dx.doi.org/10.1016/j.str.2010.08.009; PMID: 20947013
- Nelson R, Sawaya MR, Balbirnie M, Madsen AO, Riekel C, Grothe R, Eisenberg D. Structure of the cross-beta spine of amyloid-like fibrils. Nature 2005; 435:773 - 8; http://dx.doi.org/10.1038/nature03680; PMID: 15944695
- Saibil HR, Seybert A, Habermann A, Winkler J, Eltsov M, Perkovic M, Castaño-Diez D, Scheffer MP, Haselmann U, Chlanda P, et al. Heritable yeast prions have a highly organized three-dimensional architecture with interfiber structures. Proc Natl Acad Sci U S A 2012; 109:14906 - 11; http://dx.doi.org/10.1073/pnas.1211976109; PMID: 22927413
- Shewmaker F, McGlinchey RP, Wickner RB. Structural insights into functional and pathological amyloid. J Biol Chem 2011; 286:16533 - 40; http://dx.doi.org/10.1074/jbc.R111.227108; PMID: 21454545
- Wang X, Chapman MR. Curli provide the template for understanding controlled amyloid propagation. Prion 2008; 2:57 - 60; http://dx.doi.org/10.4161/pri.2.2.6746; PMID: 19098444
- Chiti F, Dobson CM. Protein misfolding, functional amyloid, and human disease. Annu Rev Biochem 2006; 75:333 - 66; http://dx.doi.org/10.1146/annurev.biochem.75.101304.123901; PMID: 16756495
- Sarell CJ, Stockley PG, Radford SE. Assessing the causes and consequences of co-polymerization in amyloid formation. Prion 2013; 7:7; http://dx.doi.org/10.4161/pri.26415; PMID: 24025483
- Derkatch IL, Uptain SM, Outeiro TF, Krishnan R, Lindquist SL, Liebman SW. Effects of Q/N-rich, polyQ, and non-polyQ amyloids on the de novo formation of the [PSI+] prion in yeast and aggregation of Sup35 in vitro. Proc Natl Acad Sci U S A 2004; 101:12934 - 9; http://dx.doi.org/10.1073/pnas.0404968101; PMID: 15326312
- Ross ED, Maclea KS, Anderson C, Ben-Hur A. A bioinformatics method for identifying Q/N-rich prion-like domains in proteins. Methods Mol Biol 2013; 1017:219 - 28; http://dx.doi.org/10.1007/978-1-62703-438-8_16; PMID: 23719919
- Kryndushkin D, Pripuzova N, Burnett BG, Shewmaker F. Non-targeted identification of prions and amyloid-forming proteins from yeast and mammalian cells. J Biol Chem 2013; 288:27100 - 11; http://dx.doi.org/10.1074/jbc.M113.485359; PMID: 23926098
- Liebman SW, Chernoff YO. Prions in yeast. Genetics 2012; 191:1041 - 72; http://dx.doi.org/10.1534/genetics.111.137760; PMID: 22879407
- McGlinchey RP, Kryndushkin D, Wickner RB. Suicidal [PSI+] is a lethal yeast prion. Proc Natl Acad Sci U S A 2011; 108:5337 - 41; http://dx.doi.org/10.1073/pnas.1102762108; PMID: 21402947
- Paushkin SV, Kushnirov VV, Smirnov VN, Ter-Avanesyan MD. Propagation of the yeast prion-like [psi+] determinant is mediated by oligomerization of the SUP35-encoded polypeptide chain release factor. EMBO J 1996; 15:3127 - 34; PMID: 8670813
- Balbirnie M, Grothe R, Eisenberg DS. An amyloid-forming peptide from the yeast prion Sup35 reveals a dehydrated beta-sheet structure for amyloid. Proc Natl Acad Sci U S A 2001; 98:2375 - 80; http://dx.doi.org/10.1073/pnas.041617698; PMID: 11226247
- Li LB, Xu K, Bonini NM. Suppression of polyglutamine toxicity by the yeast Sup35 prion domain in Drosophila. J Biol Chem 2007; 282:37694 - 701; http://dx.doi.org/10.1074/jbc.M705211200; PMID: 17956866
- Kryndushkin DS, Alexandrov IM, Ter-Avanesyan MD, Kushnirov VV. Yeast [PSI+] prion aggregates are formed by small Sup35 polymers fragmented by Hsp104. J Biol Chem 2003; 278:49636 - 43; http://dx.doi.org/10.1074/jbc.M307996200; PMID: 14507919
- Saupe SJ. A short history of small s: a prion of the fungus Podospora anserina. Prion 2007; 1:110 - 5; http://dx.doi.org/10.4161/pri.1.2.4666; PMID: 19164916
- Turcotte C, Roux A, Beauregard PB, Guérin R, Sénéchal P, Hajjar F, Rokeach LA. The calnexin-independent state does not compensate for all calnexin functions in Schizosaccharomyces pombe. FEMS Yeast Res 2007; 7:196 - 208; http://dx.doi.org/10.1111/j.1567-1364.2006.00145.x; PMID: 17328741
- Silar P, Haedens V, Rossignol M, Lalucque H. Propagation of a novel cytoplasmic, infectious and deleterious determinant is controlled by translational accuracy in Podospora anserina. Genetics 1999; 151:87 - 95; PMID: 9872950
- Wickner RB, Kryndushkin D, Shewmaker F, McGlinchey R, Edskes HK. Study of amyloids using yeast. Methods Mol Biol 2012; 849:321 - 46; http://dx.doi.org/10.1007/978-1-61779-551-0_22; PMID: 22528100
- Halfmann R, Jarosz DF, Jones SK, Chang A, Lancaster AK, Lindquist S. Prions are a common mechanism for phenotypic inheritance in wild yeasts. Nature 2012; 482:363 - 8; http://dx.doi.org/10.1038/nature10875; PMID: 22337056
- Walther TC, Brickner JH, Aguilar PS, Bernales S, Pantoja C, Walter P. Eisosomes mark static sites of endocytosis. Nature 2006; 439:998 - 1003; http://dx.doi.org/10.1038/nature04472; PMID: 16496001
- Deng C, Xiong X, Krutchinsky AN. Unifying fluorescence microscopy and mass spectrometry for studying protein complexes in cells. Mol Cell Proteomics 2009; 8:1413 - 23; http://dx.doi.org/10.1074/mcp.M800397-MCP200; PMID: 19269952
- Kelly AC, Wickner RB. Saccharomyces cerevisiae: a sexy yeast with a prion problem. Prion 2013; 7:215 - 20; http://dx.doi.org/10.4161/pri.24845; PMID: 23764836
- Tyedmers J, Madariaga ML, Lindquist S. Prion switching in response to environmental stress. PLoS Biol 2008; 6:e294; http://dx.doi.org/10.1371/journal.pbio.0060294; PMID: 19067491
- Halfmann R, Lindquist S. Epigenetics in the extreme: prions and the inheritance of environmentally acquired traits. Science 2010; 330:629 - 32; http://dx.doi.org/10.1126/science.1191081; PMID: 21030648
- Allen KD, Wegrzyn RD, Chernova TA, Müller S, Newnam GP, Winslett PA, Wittich KB, Wilkinson KD, Chernoff YO. Hsp70 chaperones as modulators of prion life cycle: novel effects of Ssa and Ssb on the Saccharomyces cerevisiae prion [PSI+]. [PSI+] Genetics 2005; 169:1227 - 42; http://dx.doi.org/10.1534/genetics.104.037168; PMID: 15545639
- Kelly AC, Shewmaker FP, Kryndushkin D, Wickner RB. Sex, prions, and plasmids in yeast. Proceedings of the National Academy of Sciences of the United States of America 2012.
- Shewmaker F, Wickner RB, Tycko R. Amyloid of the prion domain of Sup35p has an in-register parallel beta-sheet structure. Proc Natl Acad Sci U S A 2006; 103:19754 - 9; http://dx.doi.org/10.1073/pnas.0609638103; PMID: 17170131
- Bagriantsev SN, Gracheva EO, Richmond JE, Liebman SW. Variant-specific [PSI+] infection is transmitted by Sup35 polymers within [PSI+] aggregates with heterogeneous protein composition. Mol Biol Cell 2008; 19:2433 - 43; http://dx.doi.org/10.1091/mbc.E08-01-0078; PMID: 18353968
- Lopez N, Aron R, Craig EA. Specificity of class II Hsp40 Sis1 in maintenance of yeast prion [RNQ+]. [RNQ+] Mol Biol Cell 2003; 14:1172 - 81; http://dx.doi.org/10.1091/mbc.E02-09-0593; PMID: 12631732
- Ratovitski T, Chighladze E, Arbez N, Boronina T, Herbrich S, Cole RN, Ross CA. Huntingtin protein interactions altered by polyglutamine expansion as determined by quantitative proteomic analysis. Cell Cycle 2012; 11:2006 - 21; http://dx.doi.org/10.4161/cc.20423; PMID: 22580459
- Xu G, Stevens SM Jr., Moore BD, McClung S, Borchelt DR. Cytosolic proteins lose solubility as amyloid deposits in a transgenic mouse model of Alzheimer-type amyloidosis. Hum Mol Genet 2013; 22:2765 - 74; http://dx.doi.org/10.1093/hmg/ddt121; PMID: 23512986
- Gunawardena S, Her LS, Brusch RG, Laymon RA, Niesman IR, Gordesky-Gold B, Sintasath L, Bonini NM, Goldstein LS. Disruption of axonal transport by loss of huntingtin or expression of pathogenic polyQ proteins in Drosophila. Neuron 2003; 40:25 - 40; http://dx.doi.org/10.1016/S0896-6273(03)00594-4; PMID: 14527431
- Nucifora FC Jr., Ellerby LM, Wellington CL, Wood JD, Herring WJ, Sawa A, Hayden MR, Dawson VL, Dawson TM, Ross CA. Nuclear localization of a non-caspase truncation product of atrophin-1, with an expanded polyglutamine repeat, increases cellular toxicity. J Biol Chem 2003; 278:13047 - 55; http://dx.doi.org/10.1074/jbc.M211224200; PMID: 12464607