
Abstract
There is a growing body of evidence indicating that number of human neurodegenerative diseases, including Alzheimer disease, Parkinson disease, fronto-temporal dementias, and amyotrophic lateral sclerosis, propagate in the brain via prion-like intercellular induction of protein misfolding. Prions cause lethal neurodegenerative diseases in humans, the most prevalent being sporadic Creutzfeldt-Jakob disease (sCJD); they self-replicate and spread by converting the cellular form of prion protein (PrPC) to a misfolded pathogenic conformer (PrPSc). The extensive phenotypic heterogeneity of human prion diseases is determined by polymorphisms in the prion protein gene, and by prion strain-specific conformation of PrPSc. Remarkably, even though informative nucleic acid is absent, prions may undergo rapid adaptation and evolution in cloned cells and upon crossing the species barrier. In the course of our investigation of this process, we isolated distinct populations of PrPSc particles that frequently co-exist in sCJD. The human prion particles replicate independently and undergo competitive selection of those with lower initial conformational stability. Exposed to mutant substrate, the winning PrPSc conformers are subject to further evolution by natural selection of the subpopulation with the highest replication rate due to the lowest stability. Thus, the evolution and adaptation of human prions is enabled by a dynamic collection of distinct populations of particles, whose evolution is governed by the selection of progressively less stable, faster replicating PrPSc conformers. This fundamental biological mechanism may explain the drug resistance that some prions gained after exposure to compounds targeting PrPSc. Whether the phenotypic heterogeneity of other neurodegenerative diseases caused by protein misfolding is determined by the spectrum of misfolded conformers (strains) remains to be established. However, the prospect that these conformers may evolve and adapt by a prion-like mechanism calls for the reevaluation of therapeutic strategies that target aggregates of misfolded proteins, and argues for new therapeutic approaches that will focus on prior pathogenetic steps.
Broad Range of Human Prions
The human prion diseases are unique in that a single pathologic process may present as a sporadic, genetic, or infectious disease. The most common form of human prion disease, originally described as transmissible spongiform encephalopathy (TSEs), is sporadic Creutzfeldt-Jakob disease (sCJD), accounting for ~85% of cases. First shown to be transmissible to non-human primates in 1967,Citation1,Citation2 its origin and pathogenesis remain enigmatic. Now, the generally accepted model posits that the infectious pathogen responsible for TSEs is a misfolded protein, designated PrPSc.Citation3 This protein is a pathogenic conformational isoform of the normal cellular prion protein,Citation4-Citation8 PrPC, that is encoded by the host’s PRNP gene and expressed at different levels in all mammalian cells.Citation9 The discovery that misfolded proteins may be infectious represents a new biological paradigm, and although originally deemed heretical, this protein-only model is now supported by a wealth of biochemical, genetic, and animal studies,Citation5,Citation6,Citation10-Citation13 including recent success in generating infectious prions in vitro.Citation14-Citation20 The PrPSc conformer is believed to self-replicate by binding to monomers of PrPC that have predominantly α-helical secondary structure; this causes the protein to convert to the oligomeric, amyloid-forming PrPSc state with predominantly β sheet secondary structure. However, the exact structural intermediate steps remain poorly understood.Citation21,Citation22 Compared with PrPC, the β sheet secondary structure of brain-derived PrPSc increases from ~3% to ~45%, and this conformational transition leads to its insolubility in non-denaturing detergents and increased resistance to proteolysis.Citation7,Citation8 Consequently, the half-life of the protein increases from physiological ~18 h for PrPC to 36 h for PrPSc,Citation23 leading to the progressive accumulation of PrPSc in the infected brain. The lasting mystery surrounding replication of the PrPSc conformer is one of the fundamental problems of biology that remains to be solved. Two fundamental characteristics of human prion diseases are (1) the age dependency of their occurrence and (2) the extraordinary heterogeneity of the clinico-pathological phenotype.Citation24,Citation25
Human Prion Strains and Prion Coexistence
The human prion diseases are probably the most phenotypically diverse neurodegenerative disorders. The broad phenotypic heterogeneity of sporadic Creutzfeldt-Jakob disease (sCJD)Citation24 is currently understood as a complex interplay between polymorphisms in codon 129 of the PRNP gene translated to either methionine (M) or valine (V), and different PrPSc conformers coding for distinct strains of prions.Citation24,Citation26 On serial passages in the same host, distinct prion strains propagate and replicate unique phenotypes of the diseases with remarkable reproducibility, including incubation time, symptoms, distribution of pathology in the brain, and major molecular characteristics of PrPSc. Experiments in transgenic mice expressing PrPC of different species led to the conclusion that species of prion is dictated by the amino acid sequence of the host’s prion protein, and the mismatch between amino acid sequences of infecting prion and host PrPC is responsible for the so-called species barrier, which may restrict prion replication and cause a change in prion characteristics.Citation26 Variations within the same species of prion, which cause remarkably different disease phenotypes in the same host, are referred to as prion strains.Citation26,Citation27 The existence of distinct prion strains that can be passaged indefinitely was long offered as an argument for the existence of a prion-specific genome and has divided the scientific community. Subsequently, rapid progress in the past decade has produced multiple lines of evidence convincingly demonstrating that prion strain characteristics are encoded in the unique self-replicating conformation of PrPSc.Citation28-Citation31
In contrast with other mammalian prions, proteinase K digestion in sCJD prions leads to a more complex pattern with either Type 1 or Type 2 rPrPSc. These two types differ in the proteinase K cleavage site at residues ~82 (Type 1) or ~97 (Type 2), and respectively, lead to 21 or 19 kDa mass of unglycosylated fragment of protease-resistant pathogenic prion protein (rPrPSc). Using sensitive biophysical techniques, we recently discovered a broad spectrum of distinct PrPSc conformers in 20 cases of sCJD with the same codon 129 polymorphism (MM) and the same Type of rPrPSc . These data implied that sCJD is caused by a broad array of distinct prions.Citation27,Citation32,Citation33 Subsequent experiments with sedimentation velocity separation using high speed centrifugation in sucrose gradient revealed that sCJD prions exist in the continuum of particles composed of <20 to >600 PrPSc molecules. The Type 1 PrPSc particles sedimented significantly more slowly than Type 2, indicating that Type 2 PrPSc formed larger assemblies.Citation32 These findings suggest that the packing of PrPSc monomers with different conformations in distinct particles is responsible for the peptide fragmentation pattern, consisting of predominantly 19 kDa fragments in Type 2 rPrPSc, or 21 kDa in Type 1 rPrPSc, after proteinase K treatment.Citation34 Remarkably, progression rates of the disease correlate with the replication rate of human prions in vitro, which is in turn governed by the size and conformational instability of particles formed by PrPSc.Citation26,Citation32,Citation33 Cumulatively, smaller prions particles composed of less stable conformers of PrPSc replicate faster in vitro as well as in vivo.
Our subsequent experiments with highly sensitive conformation-dependent immunoassay (CDI)Citation35-Citation37 also demonstrated frequent, and perhaps universal, presence of both 21 kDa (Type 1) and 19 kDa (Type 2) unglycosylated fragments of protease-resistant (r) PrPSc in the same sCJD brain.Citation34 The fragments were present at different ratios, and indicate co-occurrence of markedly different PrPSc conformers, often in the same anatomical structure in the same brain. These quantitative findings extended previous qualitative observations with diverse antibodies and western blot techniques.Citation38-Citation42 Apart from challenging the validity of the clinicopathological classification of sCJD based on PRNP gene polymorphism and western blot patterns of Type 1 or Type 2 rPrPSc 24, these findings raised some fundamental questions: (1) Do the coexistent Type 1 and Type 2 rPrPSc form distinct or hybrid particles composed of both types of PrPSc? (2) What is the impact of coexistence of distinct PrPSc conformers? (3) Do they replicate independently and thus imply co-existence of different sCJD prions?
Although the possible coexistence of different prions in naturally prion-infected sheep, goat, and mink has been suspected early on,Citation28,Citation43-Citation47 the early experiments could not differentiate between two possibilities: (1) strain adaptation caused by a switch from the primary sequence of the original host’s PrPSc to a different PrP sequence in the new host, or (2) selection of strains from a co-existing pool in the natural host.Citation48,Citation49 Since Type 1 and Type 2 prion particles can be separated by sedimentation velocity,Citation32 we investigated whether coexistent Type 1 and Type 2 rPrPSc form distinct particles. Using high-speed centrifugation in sucrose gradient and sedimentation velocity separation in tandem with CDI and conformational stability assay, we isolated two populations of prion particles, each composed of a relatively homogenous population of conformers that had either Type 1 or Type 2 N-terminus proteolytic cleavage sites and different conformational stability (). Since we did not observe a change in the sedimentation velocity nor the formation of hybrids after mixing isolated “pure” MM1 and MM2 rPrPSc in vitro, we concluded that they do not interact. Thus, our findings indicate that the coexistence of distinct prion particles with different conformational structure, or packing of the monomers of PrPSc, is a common feature of sCJD.
Figure 1. Schematic reaction coordinates of conformational transition from less stable PrPC to more stable PrPSc, and conformational evolution of sCJD PrPSc. The isolates of sCJD prions homozygous for methionine in codon 129 are composed of two populations of PrPSc conformers: less stable Type 1A PrPSc and more stable Type 2. Their replication with unglycosylated mutant PrPC(N181,197Q) substrate leads to initial preferential amplification of less stable Type 1A PrPSc and continuing selection of progressively less stable Type 1B PrPSc. The ΔG is the energy difference between unfolded and folded state of PrPC; ΔG# is the activation energy necessary for conformational transition from PrPC to PrPSc state.Citation50
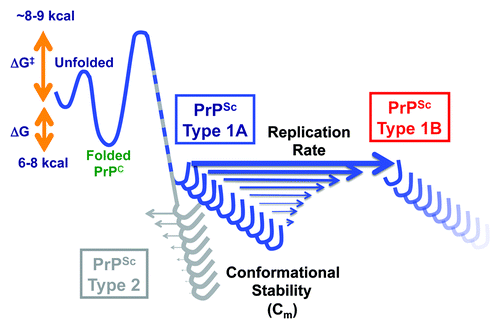
Mechanism of adaptation, evolution, and competition of prions
Change in biological characteristics of prions observed upon crossing the species barrier and in experiments with subcloned cell lines indicate that prions may undergo evolution and adaptation, but the exact molecular mechanism of this effect has remained speculative.Citation31,Citation49,Citation51 To investigate the impact of the coexistent prion particle types on this process, we recently used a modified protein misfolding cyclic amplification (PMCA)Citation52 with homologous, as well as mutant, unglycosylated PrPC(N181,197Q) substrate carrying methionine in codon 129.Citation34 The serial PMCA of “pure” Type MM1 and mixed Type MM1+2 sCJD seeds underwent two distinct phases. In the first adaptation phase, the amplification was limited, and detectable only with CDI. In the second replication phase, we observed an abrupt increase in the amplification rate. We selected for these experiments sCJD samples that had approximately equal concentrations of Type 1 and Type 2 rPrPSc. Remarkably, within the Type 1+2 particle mixture, Type 2 rPrPSc gradually disappeared, even though “pure” Type 2 sCJD amplified very well. This effect resulted in the uniform selection of Type 1 rPrPSc in mixed Type 1+2 cases and a progressive drop in the stability of the amplified rPrPSc. These findings provided the first experimental evidence for an evolutionary process within the Type 1+2 prion mixture, with selection that favors Type 1 conformers with the lowest stability.Citation34 The initial preferential amplification rate of Type 1 PrPSc is not surprising, since there is typically a higher percentage of less stable protease sensitive oligomers in Type 1 PrPSc, as found in our recent studies. This may explain why this rPrPSc subtype represents ~70% of all sCJD cases.Citation27,Citation32 Our data also correlate with the superior transmissibility and short incubation times of MM1 sCJD prions, and with incomplete transmissions and extended incubations times of MM2 sCJD prions observed in transgenic mice that overexpress homologous or chimeric human PrPC.Citation53,Citation54
The inhibition of Type 2 amplification that occurs in the Type 1+2 mixture contrasted sharply with the very efficient replication of “pure” Type 2 sCJD (with less than 5% of Type 1 present). These findings indicate that the unglycosylated PrPC(N181,197Q) is not per se a preferential substrate for amplification of Type 1 PrPSc, and suggest interference between Type 1 and Type 2 conformers during replication. The prion interference has been observed in vivo in mice and Syrian hamsters that were inoculated simultaneously or sequentially with two distinct strains of prions.Citation55-Citation58 Since we observed no direct interaction between different conformers of PrPSc in our in vitro mixing experiments with “pure” MM1 and MM2 sCJD prions, we concluded that the most likely explanation is competition for PrPC substrate or auxiliary molecule; however, the exact molecular mechanism of this phenomenon remains to be fully elucidated.Citation34
Cumulatively, the distinct particle size, conformational stability, and amplification rate of these prion subtypes argue for the frequent coexistence of different sCJD prions in the same host (). Under favorable conditions with compatible PrPC substrate, the mixture of human PrPSc conformers may undergo an evolution that selects a subset with the highest replication rate, due to the lowest stability (). Notably, the adaptation phase and prion strain evolution inferred from experiments with cloned cellsCitation31 and Tg mice,Citation48,Citation49 has been shown in our experiments to be a conformational process. Thus, the selection of a relatively narrow population of conformers with similar conformational stability during passage in experimental animals or cells, together with high activation energy barriers preventing conversion to different prion strains, is likely responsible for the exceptional stability of the biological characteristics of laboratory prion strains, as long as they are propagated in the same host or cells. However, many important questions remain unanswered, specifically, (1) how the initial ratio between Type 1 and Type 2 would influence the outcome, and (2) whether the first adaptation phase is due to the requirement for the critical threshold stoichiometry between seed PrPSc and mutant substrate PrPC(N181,197Q) needed for optimum replication, or (3) whether adaptation is due to the absence of sugar chains on PrPC(N181,197Q) substrate and double N→Q mutation, or (4) whether this first phase is caused by the difference between mouse and human auxiliary molecules. Since all our PMCA experiments were performed with PrPs carrying methionine in codon 129 of the PRNP gene, it also remains to be established if valine in the same position will impact the process. However, the evolutionary conformational selection mechanism of PrPSc may explain the recently observed drug-induced evolution of mammalian prions.Citation59 In these experiments, Oelschlegel and Weissmann exposed different prion-infected cell sub-lines to the drug (swainsonine) and observed not only drug-resistant, but also drug-dependent prion populations, which propagated more rapidly in the presence rather than the absence of the drug. Moreover, their data demonstrated that new, initially drug-dependent prions became new stable prion variants after drug withdrawal. These prion adaptations are most likely driven by the conformational selection mechanism we observed in our experiment in vitro and call for the reevaluation of different therapeutic strategies that target amyloid-forming aggregates of PrPSc. High-resolution structural tools and research into the role of PrPSc ligands must address the apparent conformational plasticity of PrPSc, which is likely responsible for the coexistent spectrum of prion conformers, and enables the prion evolution that results in extensive phenotypic diversity ().
Implications for neurodegenerative diseases caused by protein misfolding
The advanced understanding of clinicopathological heterogeneity and pathogenesis of late onset Alzheimer disease (AD), Parkinson disease (PD), amyotrophic lateral sclerosis (ALS), and other diseases linked to protein misfolding, demands that we identify the factors that lead to a spectrum of different phenotypes and different progression rates. The most frequent form of dementia is late-onset (>65 y of age) Alzheimer diseases (AD).Citation60 However, both early and late onset forms are pathologically characterized by the presence of amyloid β peptides (Aβ) plaques, and intraneuronal tangles of hyperphosphorylated forms of microtubule associated protein tau (MAPT).Citation61 The causal mutations in amyloid precursor protein (APP), presenilin 1 (PSEN1), and presenilin 2 (PSEN2) genes, which have been identified in early-onset forms, established the central role of amyloid β (Aβ) and its processing in AD.Citation62 However, the role of the amyloid deposits and tangles in the cognitive decline and pathogenesis of late-onset sporadic AD is more difficult to define. A major determinant in the risk of late-onset AD is the polymorphism of the APOE gene, in which a single e4 allele increases the risk by a factor of 4, and two e4 alleles increases the risk by a factor of 13. Additional polymorphisms in several recently identified genes may moderately increase the risk of disease.Citation63 Thus, the extensive phenotypic variability of AD with variable progression rates,Citation64,Citation65 clinical symptomatology,Citation66 and pathological findingsCitation67,Citation68 suggests a complex impact of different structural forms of misfolded Aβ and tau proteins,Citation69 variable genetic backgrounds,Citation63 and compensatory mechanisms (“cognitive reserves”).Citation70
While the genetic and environmental factors linked to the risk of developing AD are well recognized, the factors leading to variable progression rates of late-onset AD are unknown.Citation63 Recently, the Prion Surveillance Centers in the US and Europe independently described a novel subgroup of patients who have rapidly progressive dementia that clinically imitates prion diseases, and which, after exhausting neuropathological investigation and prion protein gene sequencing, is concluded to be rapidly progressive late-onset AD (rpAD).Citation64,Citation71-Citation74 The data collected from multiple Prion Centers uniformly demonstrate the absence of positive family history or comorbidity, the presence of distinctive clinical characteristics, and a frequency of e4 alleles in the APOE gene that corresponds to the general population. A systematic investigation of the genetics and molecular pathology of Aβ and tau in those patients should lead to the identification of biological factors responsible for the variable progression rates of AD. These findings will be crucial in developing new therapeutic targets for AD, for preclinical diagnostics, and for individualized therapeutic approaches.Citation60
Investigating the conformational structure of brain Aβ and tau is critical for deciphering their role in the variable progression rates and phenotypes of AD. Extensive analysis of aging brain samples indicates that the pathological process underlying AD starts early in isolated brain anatomical structures and then spreads through neuronal projections.Citation67 This process can be accelerated in transgenic mice models of AD and tauopathy by intracerebral injection of preformed misfolded Aβ or tau, and different structural conformers of misfolded proteins have varying potency to accelerate the pathology.Citation75,Citation76 These data suggest a prion-like mechanism, and since synthetic Aβ is significantly less active in this “seeding” effect than Aβ of brain origin, the data imply a conformational and biological plasticity, which is the basis for prion subtypes (strains).Citation26,Citation77,Citation78 These findings have raised some fundamental questions, specifically, whether different conformers of Aβ or tau contribute to varying progression rates of the disease, and whether subtle differences in the conformation of Aβ or tau may be responsible for the distinct disease phenotypes. Additionally, there is a large body of literature on the conformational characteristics and folding pathways of synthetic and recombinant Aβ and tau, which raises the question of how these variable structures are relevant to the structure of brain Aβ and tau and to the pathogenesis of the disease.
Several therapeutic trials targeting amyloid deposits in AD have failed. These disappointing results triggered a reexamination of the pathogenetic assumptions that lead to their development, and exposed a critical need for new therapeutic targets and earlier diagnostic detection of the disease.Citation79 This goal is especially important in light of our investigations of prion adaptation and evolution, which imply that misfolded proteins, including those causing AD and PD may evolve, and thus gain resistance to the therapeutic ligand that originally targeted them. Equally important is to advance our understanding of phenotypic heterogeneity in AD, and the essential requirement for the identification of genetic and conformational proteomic markers that would differentiate distinct subgroups of patients, who may respond differently to administered therapeutics.
| Abbreviations: | ||
| AD | = | Alzheimer disease |
| ALS | = | amyotrophic lateral sclerosis |
| APOE | = | apolipoprotein E gene |
| CDI | = | conformation-dependent immunoassay |
| MM | = | MV, VV common polymorphisms in codon 129 of PRNP gene coding for methionine (M) or valine (V) |
| MM1 sCJD | = | sporadic Creutzfeldt-Jakob disease homozygous for methionine (M) in polymorphic codon 129 of PRNP gene and 21 kDa fragment (Type 1) of unglycosylated rPrPSc on WBs |
| MM2 sCJD | = | sporadic Creutzfeldt-Jakob disease homozygous for methionine (M) in polymorphic codon 129 of PRNP gene and 19 kDa fragment (Type 2) of unglycosylated rPrPSc on WBs |
| PD | = | Parkinson disease |
| PRNP | = | prion protein gene |
| PrP | = | prion protein |
| PrPC | = | normal or cellular prion protein |
| PrPSc | = | misfolded pathogenic prion protein |
| rPrPSc | = | protease-resistant conformers of pathogenic prion protein (PrP 27-30) |
| sPMCA | = | serial Protein Misfolding Cyclic Amplification |
| sPrPSc | = | protease-sensitive conformers of pathogenic prion protein |
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
The authors are grateful to the patient’s families, the CJD Foundation, and all the members of the National Prion Disease Pathology Surveillance Center. This work was supported by grants from NIH (NS074317), CDC (UR8/CCU515004), and the Charles S. Britton Fund.
Related Research Data
References
- Gibbs CJ Jr., Gajdusek DC, Asher DM, Alpers MP, Beck E, Daniel PM, Matthews WB. Creutzfeldt-Jakob disease (spongiform encephalopathy): transmission to the chimpanzee. Science 1968; 161:388 - 9; http://dx.doi.org/10.1126/science.161.3839.388; PMID: 5661299
- Brown P, Gibbs CJ Jr., Rodgers-Johnson P, Asher DM, Sulima MP, Bacote A, Goldfarb LG, Gajdusek DC. Human spongiform encephalopathy: the National Institutes of Health series of 300 cases of experimentally transmitted disease. Ann Neurol 1994; 35:513 - 29; http://dx.doi.org/10.1002/ana.410350504; PMID: 8179297
- Prusiner SB. Novel proteinaceous infectious particles cause scrapie. Science 1982; 216:136 - 44; http://dx.doi.org/10.1126/science.6801762; PMID: 6801762
- Pan K-M, Baldwin M, Nguyen J, Gasset M, Serban A, Groth D, Mehlhorn I, Huang Z, Fletterick RJ, Cohen FE, et al. Conversion of α-helices into β-sheets features in the formation of the scrapie prion proteins. Proc Natl Acad Sci U S A 1993; 90:10962 - 6; http://dx.doi.org/10.1073/pnas.90.23.10962; PMID: 7902575
- Prusiner SB. Prions. Proc Natl Acad Sci U S A 1998; 95:13363 - 83; http://dx.doi.org/10.1073/pnas.95.23.13363; PMID: 9811807
- Prusiner SB, ed. Prion Biology and Diseases. Cold Spring Harbor: Cold Spring Harbor Laboratory Press, 2004.
- Safar J, Roller PP, Gajdusek DC, Gibbs CJ Jr.. Conformational transitions, dissociation, and unfolding of scrapie amyloid (prion) protein. J Biol Chem 1993; 268:20276 - 84; PMID: 8104185
- Caughey BW, Dong A, Bhat KS, Ernst D, Hayes SF, Caughey WS. Secondary structure analysis of the scrapie-associated protein PrP 27-30 in water by infrared spectroscopy. Biochemistry 1991; 30:7672 - 80; http://dx.doi.org/10.1021/bi00245a003; PMID: 1678278
- Oesch B, Westaway D, Wälchli M, McKinley MP, Kent SBH, Aebersold R, Barry RA, Tempst P, Teplow DB, Hood LE, et al. A cellular gene encodes scrapie PrP 27-30 protein. Cell 1985; 40:735 - 46; http://dx.doi.org/10.1016/0092-8674(85)90333-2; PMID: 2859120
- Caughey B, Baron GS, Chesebro B, Jeffrey M. Getting a grip on prions: oligomers, amyloids, and pathological membrane interactions. Annu Rev Biochem 2009; 78:177 - 204; http://dx.doi.org/10.1146/annurev.biochem.78.082907.145410; PMID: 19231987
- Cobb NJ, Surewicz WK. Prion diseases and their biochemical mechanisms. Biochemistry 2009; 48:2574 - 85; http://dx.doi.org/10.1021/bi900108v; PMID: 19239250
- Collinge J, Clarke AR. A general model of prion strains and their pathogenicity. Science 2007; 318:930 - 6; http://dx.doi.org/10.1126/science.1138718; PMID: 17991853
- Morales R, Abid K, Soto C. The prion strain phenomenon: molecular basis and unprecedented features. Biochim Biophys Acta 2007; 1772:681 - 91; http://dx.doi.org/10.1016/j.bbadis.2006.12.006; PMID: 17254754
- Legname G, Baskakov IV, Nguyen H-OB, Riesner D, Cohen FE, DeArmond SJ, Prusiner SB. Synthetic mammalian prions. Science 2004; 305:673 - 6; http://dx.doi.org/10.1126/science.1100195; PMID: 15286374
- Castilla J, Saá P, Hetz C, Soto C. In vitro generation of infectious scrapie prions. Cell 2005; 121:195 - 206; http://dx.doi.org/10.1016/j.cell.2005.02.011; PMID: 15851027
- Barria MA, Mukherjee A, Gonzalez-Romero D, Morales R, Soto C. De novo generation of infectious prions in vitro produces a new disease phenotype. PLoS Pathog 2009; 5:e1000421; http://dx.doi.org/10.1371/journal.ppat.1000421; PMID: 19436715
- Wang F, Wang X, Yuan CG, Ma J. Generating a prion with bacterially expressed recombinant prion protein. Science 2010; 327:1132 - 5; http://dx.doi.org/10.1126/science.1183748; PMID: 20110469
- Deleault NR, Harris BT, Rees JR, Supattapone S. Formation of native prions from minimal components in vitro. Proc Natl Acad Sci U S A 2007; 104:9741 - 6; http://dx.doi.org/10.1073/pnas.0702662104; PMID: 17535913
- Geoghegan JC, Miller MB, Kwak AH, Harris BT, Supattapone S. Trans-dominant inhibition of prion propagation in vitro is not mediated by an accessory cofactor. PLoS Pathog 2009; 5:e1000535; http://dx.doi.org/10.1371/journal.ppat.1000535; PMID: 19649330
- Kim JI, Cali I, Surewicz K, Kong Q, Raymond GJ, Atarashi R, Race B, Qing L, Gambetti P, Caughey B, et al. Mammalian prions generated from bacterially expressed prion protein in the absence of any mammalian cofactors. J Biol Chem 2010; 285:14083 - 7; http://dx.doi.org/10.1074/jbc.C110.113464; PMID: 20304915
- Kocisko DA, Come JH, Priola SA, Chesebro B, Raymond GJ, Lansbury PT Jr., Caughey B. Cell-free formation of protease-resistant prion protein. Nature 1994; 370:471 - 4; http://dx.doi.org/10.1038/370471a0; PMID: 7913989
- Prusiner SB. Prion diseases and the BSE crisis. Science 1997; 278:245 - 51; http://dx.doi.org/10.1126/science.278.5336.245; PMID: 9323196
- Safar JG, DeArmond SJ, Kociuba K, Deering C, Didorenko S, Bouzamondo-Bernstein E, Prusiner SB, Tremblay P. Prion clearance in bigenic mice. J Gen Virol 2005; 86:2913 - 23; http://dx.doi.org/10.1099/vir.0.80947-0; PMID: 16186247
- Puoti G, Bizzi A, Forloni G, Safar JG, Tagliavini F, Gambetti P. Sporadic human prion diseases: molecular insights and diagnosis. Lancet Neurol 2012; 11:618 - 28; http://dx.doi.org/10.1016/S1474-4422(12)70063-7; PMID: 22710755
- Parchi P, de Boni L, Saverioni D, Cohen ML, Ferrer I, Gambetti P, Gelpi E, Giaccone G, Hauw JJ, Höftberger R, et al. Consensus classification of human prion disease histotypes allows reliable identification of molecular subtypes: an inter-rater study among surveillance centres in Europe and USA. Acta Neuropathol 2012; 124:517 - 29; http://dx.doi.org/10.1007/s00401-012-1002-8; PMID: 22744790
- Safar JG. Molecular Mechanisms Encoding Quantitative and Qualitative Traits of Prion Strains. In: Gambetti P, ed. Prions and Diseases. New York: Springer Verlag, 2012.
- Safar JG. Molecular pathogenesis of sporadic prion diseases in man. Prion 2012; 6:108 - 15; http://dx.doi.org/10.4161/pri.18666; PMID: 22421210
- Bessen RA, Marsh RF. Distinct PrP properties suggest the molecular basis of strain variation in transmissible mink encephalopathy. J Virol 1994; 68:7859 - 68; PMID: 7966576
- Telling GC, Parchi P, DeArmond SJ, Cortelli P, Montagna P, Gabizon R, Mastrianni J, Lugaresi E, Gambetti P, Prusiner SB. Evidence for the conformation of the pathologic isoform of the prion protein enciphering and propagating prion diversity. Science 1996; 274:2079 - 82; http://dx.doi.org/10.1126/science.274.5295.2079; PMID: 8953038
- Safar J, Prusiner SB. Molecular studies of prion diseases. Prog Brain Res 1998; 117:421 - 34; http://dx.doi.org/10.1016/S0079-6123(08)64030-3; PMID: 9932423
- Li J, Browning S, Mahal SP, Oelschlegel AM, Weissmann C. Darwinian evolution of prions in cell culture. Science 2010; 327:869 - 72; http://dx.doi.org/10.1126/science.1183218; PMID: 20044542
- Kim C, Haldiman T, Surewicz K, Cohen Y, Chen W, Blevins J, Sy MS, Cohen M, Kong Q, Telling GC, et al. Small protease sensitive oligomers of PrPSc in distinct human prions determine conversion rate of PrP(C). PLoS Pathog 2012; 8:e1002835; http://dx.doi.org/10.1371/journal.ppat.1002835; PMID: 22876179
- Kim C, Haldiman T, Cohen Y, Chen W, Blevins J, Sy MS, Cohen M, Safar JG. Protease-sensitive conformers in broad spectrum of distinct PrPSc structures in sporadic Creutzfeldt-Jakob disease are indicator of progression rate. PLoS Pathog 2011; 7:e1002242; http://dx.doi.org/10.1371/journal.ppat.1002242; PMID: 21931554
- Haldiman T, Kim C, Cohen Y, Chen W, Blevins J, Qing L, Cohen ML, Langeveld J, Telling GC, Kong Q, et al. Co-existence of distinct prion types enables conformational evolution of human PrPSc by competitive selection. J Biol Chem 2013; 288:29846 - 61; http://dx.doi.org/10.1074/jbc.M113.500108; PMID: 23974118
- Safar JG, Geschwind MD, Deering C, Didorenko S, Sattavat M, Sanchez H, Serban A, Vey M, Baron H, Giles K, et al. Diagnosis of human prion disease. Proc Natl Acad Sci U S A 2005; 102:3501 - 6; http://dx.doi.org/10.1073/pnas.0409651102; PMID: 15741275
- Safar JG, Scott M, Monaghan J, Deering C, Didorenko S, Vergara J, Ball H, Legname G, Leclerc E, Solforosi L, et al. Measuring prions causing bovine spongiform encephalopathy or chronic wasting disease by immunoassays and transgenic mice. Nat Biotechnol 2002; 20:1147 - 50; http://dx.doi.org/10.1038/nbt748; PMID: 12389035
- Safar J, Wille H, Itri V, Groth D, Serban H, Torchia M, Cohen FE, Prusiner SB. Eight prion strains have PrP(Sc) molecules with different conformations. Nat Med 1998; 4:1157 - 65; http://dx.doi.org/10.1038/2654; PMID: 9771749
- Uro-Coste E, Cassard H, Simon S, Lugan S, Bilheude JM, Perret-Liaudet A, Ironside JW, Haik S, Basset-Leobon C, Lacroux C, et al. Beyond PrP9res) type 1/type 2 dichotomy in Creutzfeldt-Jakob disease. PLoS Pathog 2008; 4:e1000029; http://dx.doi.org/10.1371/journal.ppat.1000029; PMID: 18389084
- Yull HM, Ritchie DL, Langeveld JP, van Zijderveld FG, Bruce ME, Ironside JW, Head MW. Detection of type 1 prion protein in variant Creutzfeldt-Jakob disease. Am J Pathol 2006; 168:151 - 7; http://dx.doi.org/10.2353/ajpath.2006.050766; PMID: 16400018
- Schoch G, Seeger H, Bogousslavsky J, Tolnay M, Janzer RC, Aguzzi A, Glatzel M. Analysis of prion strains by PrPSc profiling in sporadic Creutzfeldt-Jakob disease. PLoS Med 2006; 3:e14; http://dx.doi.org/10.1371/journal.pmed.0030014; PMID: 16354106
- Cali I, Castellani R, Alshekhlee A, Cohen Y, Blevins J, Yuan J, Langeveld JP, Parchi P, Safar JG, Zou WQ, et al. Co-existence of scrapie prion protein types 1 and 2 in sporadic Creutzfeldt-Jakob disease: its effect on the phenotype and prion-type characteristics. Brain 2009; 132:2643 - 58; http://dx.doi.org/10.1093/brain/awp196; PMID: 19734292
- Kobayashi A, Mizukoshi K, Iwasaki Y, Miyata H, Yoshida Y, Kitamoto T. Co-occurrence of types 1 and 2 PrP(res) in sporadic Creutzfeldt-Jakob disease MM1. Am J Pathol 2011; 178:1309 - 15; http://dx.doi.org/10.1016/j.ajpath.2010.11.069; PMID: 21356381
- Kimberlin RH, Walker CA. Evidence that the transmission of one source of scrapie agent to hamsters involves separation of agent strains from a mixture. J Gen Virol 1978; 39:487 - 96; http://dx.doi.org/10.1099/0022-1317-39-3-487; PMID: 96212
- Bruce ME, Fraser H. Scrapie strain variation and its implications. Curr Top Microbiol Immunol 1991; 172:125 - 38; http://dx.doi.org/10.1007/978-3-642-76540-7_8; PMID: 1810707
- Bruce ME. Scrapie strain variation and mutation. Br Med Bull 1993; 49:822 - 38; PMID: 8137131
- Bessen RA, Marsh RF. Identification of two biologically distinct strains of transmissible mink encephalopathy in hamsters. J Gen Virol 1992; 73:329 - 34; http://dx.doi.org/10.1099/0022-1317-73-2-329; PMID: 1531675
- Bessen RA, Marsh RF. Biochemical and physical properties of the prion protein from two strains of the transmissible mink encephalopathy agent. J Virol 1992; 66:2096 - 101; PMID: 1347795
- Scott MR, Peretz D, Nguyen H-OB, Dearmond SJ, Prusiner SB. Transmission barriers for bovine, ovine, and human prions in transgenic mice. J Virol 2005; 79:5259 - 71; http://dx.doi.org/10.1128/JVI.79.9.5259-5271.2005; PMID: 15827140
- Scott M, Peretz D, Ridley RM, Baker HF, DeArmond SJ, Prusiner SB. Transgenetic investigations of the species barrier and prion strains. In: Prusiner SB, ed. Prion Biology and Diseases. Cold Spring Harbor: Cold Spring Harbor Laboratory Press, 2004:435-82.
- Govaerts C, Wille H, Prusiner SB, Cohen FE. Structural studies of prion proteins. In: Prusiner SB, ed. Prion Biology and Diseases. Cold Spring Harbor: Cold Spring Harbor Laboratory Press, 2004:243-82.
- Mahal SP, Baker CA, Demczyk CA, Smith EW, Julius C, Weissmann C. Prion strain discrimination in cell culture: the cell panel assay. Proc Natl Acad Sci U S A 2007; 104:20908 - 13; http://dx.doi.org/10.1073/pnas.0710054104; PMID: 18077360
- Castilla J, Morales R, Saá P, Barria M, Gambetti P, Soto C. Cell-free propagation of prion strains. EMBO J 2008; 27:2557 - 66; http://dx.doi.org/10.1038/emboj.2008.181; PMID: 18800058
- Bishop MT, Will RG, Manson JC. Defining sporadic Creutzfeldt-Jakob disease strains and their transmission properties. Proc Natl Acad Sci U S A 2010; 107:12005 - 10; http://dx.doi.org/10.1073/pnas.1004688107; PMID: 20547859
- Korth C, Kaneko K, Groth D, Heye N, Telling G, Mastrianni J, Parchi P, Gambetti P, Will R, Ironside J, et al. Abbreviated incubation times for human prions in mice expressing a chimeric mouse-human prion protein transgene. Proc Natl Acad Sci U S A 2003; 100:4784 - 9; http://dx.doi.org/10.1073/pnas.2627989100; PMID: 12684540
- Bartz JC, Kramer ML, Sheehan MH, Hutter JA, Ayers JI, Bessen RA, Kincaid AE. Prion interference is due to a reduction in strain-specific PrPSc levels. J Virol 2007; 81:689 - 97; http://dx.doi.org/10.1128/JVI.01751-06; PMID: 17079313
- Bartz JC, Aiken JM, Bessen RA. Delay in onset of prion disease for the HY strain of transmissible mink encephalopathy as a result of prior peripheral inoculation with the replication-deficient DY strain. J Gen Virol 2004; 85:265 - 73; http://dx.doi.org/10.1099/vir.0.19394-0; PMID: 14718642
- Shikiya RA, Ayers JI, Schutt CR, Kincaid AE, Bartz JC. Coinfecting prion strains compete for a limiting cellular resource. J Virol 2010; 84:5706 - 14; http://dx.doi.org/10.1128/JVI.00243-10; PMID: 20237082
- Manuelidis L, Lu ZY. Virus-like interference in the latency and prevention of Creutzfeldt-Jakob disease. Proc Natl Acad Sci U S A 2003; 100:5360 - 5; http://dx.doi.org/10.1073/pnas.0931192100; PMID: 12692308
- Oelschlegel AM, Weissmann C. Acquisition of drug resistance and dependence by prions. PLoS Pathog 2013; 9:e1003158; http://dx.doi.org/10.1371/journal.ppat.1003158; PMID: 23408888
- Cummings JL. Biomarkers in Alzheimer’s disease drug development. Alzheimers Dement 2011; 7:e13 - 44; http://dx.doi.org/10.1016/j.jalz.2010.06.004; PMID: 21550318
- Braak H, Thal DR, Ghebremedhin E, Del Tredici K. Stages of the pathologic process in Alzheimer disease: age categories from 1 to 100 years. J Neuropathol Exp Neurol 2011; 70:960 - 9; http://dx.doi.org/10.1097/NEN.0b013e318232a379; PMID: 22002422
- Hardy J. Has the amyloid cascade hypothesis for Alzheimer’s disease been proved?. Curr Alzheimer Res 2006; 3:71 - 3; http://dx.doi.org/10.2174/156720506775697098; PMID: 16472206
- Schellenberg GD, Montine TJ. The genetics and neuropathology of Alzheimer’s disease. Acta Neuropathol 2012; 124:305 - 23; http://dx.doi.org/10.1007/s00401-012-0996-2; PMID: 22618995
- Schmidt C, Wolff M, Weitz M, Bartlau T, Korth C, Zerr I. Rapidly progressive Alzheimer disease. Arch Neurol 2011; 68:1124 - 30; http://dx.doi.org/10.1001/archneurol.2011.189; PMID: 21911694
- Wilkosz PA, Seltman HJ, Devlin B, Weamer EA, Lopez OL, DeKosky ST, Sweet RA. Trajectories of cognitive decline in Alzheimer’s disease. Int Psychogeriatr 2010; 22:281 - 90; http://dx.doi.org/10.1017/S1041610209991001; PMID: 19781112
- McKhann GM, Knopman DS, Chertkow H, Hyman BT, Jack CR Jr., Kawas CH, Klunk WE, Koroshetz WJ, Manly JJ, Mayeux R, et al. The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement 2011; 7:263 - 9; http://dx.doi.org/10.1016/j.jalz.2011.03.005; PMID: 21514250
- Braak H, Del Tredici K. Evolutional aspects of Alzheimer’s disease pathogenesis. J Alzheimers Dis 2013; 33:Suppl 1 S155 - 61; PMID: 22699850
- Weiner MW, Veitch DP, Aisen PS, Beckett LA, Cairns NJ, Green RC, Harvey D, Jack CR, Jagust W, Liu E, et al, Alzheimer’s Disease Neuroimaging Initiative. The Alzheimer’s Disease Neuroimaging Initiative: a review of papers published since its inception. Alzheimers Dement 2012; 8:Suppl S1 - 68; http://dx.doi.org/10.1016/j.jalz.2011.09.172; PMID: 22047634
- Ashe KH, Aguzzi A. Prions, prionoids and pathogenic proteins in Alzheimer disease. Prion 2013; 7:55 - 9; http://dx.doi.org/10.4161/pri.23061; PMID: 23208281
- Nelson PT, Alafuzoff I, Bigio EH, Bouras C, Braak H, Cairns NJ, Castellani RJ, Crain BJ, Davies P, Del Tredici K, et al. Correlation of Alzheimer disease neuropathologic changes with cognitive status: a review of the literature. J Neuropathol Exp Neurol 2012; 71:362 - 81; http://dx.doi.org/10.1097/NEN.0b013e31825018f7; PMID: 22487856
- Schmidt C, Artjomova S, Hoeschel M, Zerr I. CSF prion protein concentration and cognition in patients with Alzheimer disease. Prion 2013; 7:229 - 34; http://dx.doi.org/10.4161/pri.23904; PMID: 23406922
- Schmidt C, Haïk S, Satoh K, Rábano A, Martinez-Martin P, Roeber S, Brandel JP, Calero-Lara M, de Pedro-Cuesta J, Laplanche JL, et al. Rapidly progressive Alzheimer’s disease: a multicenter update. J Alzheimers Dis 2012; 30:751 - 6; PMID: 22460329
- Schmidt C, Redyk K, Meissner B, Krack L, von Ahsen N, Roeber S, Kretzschmar H, Zerr I. Clinical features of rapidly progressive Alzheimer’s disease. Dement Geriatr Cogn Disord 2010; 29:371 - 8; http://dx.doi.org/10.1159/000278692; PMID: 20453509
- Chitravas N, Jung RS, Kofskey DM, Blevins JE, Gambetti P, Leigh RJ, Cohen ML. Treatable neurological disorders misdiagnosed as Creutzfeldt-Jakob disease. Ann Neurol 2011; 70:437 - 44; http://dx.doi.org/10.1002/ana.22454; PMID: 21674591
- Kane MD, Lipinski WJ, Callahan MJ, Bian F, Durham RA, Schwarz RD, Roher AE, Walker LC. Evidence for seeding of beta -amyloid by intracerebral infusion of Alzheimer brain extracts in beta -amyloid precursor protein-transgenic mice. J Neurosci 2000; 20:3606 - 11; PMID: 10804202
- Guo JL, Lee VM. Neurofibrillary tangle-like tau pathology induced by synthetic tau fibrils in primary neurons over-expressing mutant tau. FEBS Lett 2013; 587:717 - 23; http://dx.doi.org/10.1016/j.febslet.2013.01.051; PMID: 23395797
- Meyer-Luehmann M, Coomaraswamy J, Bolmont T, Kaeser S, Schaefer C, Kilger E, Neuenschwander A, Abramowski D, Frey P, Jaton AL, et al. Exogenous induction of cerebral beta-amyloidogenesis is governed by agent and host. Science 2006; 313:1781 - 4; http://dx.doi.org/10.1126/science.1131864; PMID: 16990547
- Prusiner SB. Cell biology. A unifying role for prions in neurodegenerative diseases. Science 2012; 336:1511 - 3; http://dx.doi.org/10.1126/science.1222951; PMID: 22723400
- Colom LV, Perry G, Kuljis RO. Tackling the elusive challenges relevant to conquering the 100-plus year old problem of Alzheimer’s disease. Curr Alzheimer Res 2013; 10:108 - 16; PMID: 23368433