
Abstract
The tumor suppressor protein p53 loses its function in more than 50% of human malignant tumors. Recent studies have suggested that mutant p53 can form aggregates that are related to loss-of-function effects, negative dominance and gain-of-function effects and cancers with a worsened prognosis. In recent years, several degenerative diseases have been shown to have prion-like properties similar to mammalian prion proteins (PrPs). However, whereas prion diseases are rare, the incidence of these neurodegenerative pathologies is high. Malignant tumors involving mutated forms of the tumor suppressor p53 protein seem to have similar substrata. The aggregation of the entire p53 protein and three functional domains of p53 into amyloid oligomers and fibrils has been demonstrated. Amyloid aggregates of mutant p53 have been detected in breast cancer and malignant skin tumors. Most p53 mutations related to cancer development are found in the DNA-binding domain (p53C), which has been experimentally shown to form amyloid oligomers and fibrils. Several computation programs have corroborated the predicted propensity of p53C to form aggregates, and some of these programs suggest that p53C is more likely to form aggregates than the globular domain of PrP. Overall, studies imply that mutant p53 exerts a dominant-negative regulatory effect on wild-type (WT) p53 and exerts gain-of-function effects when co-aggregating with other proteins such as p63, p73 and acetyltransferase p300. We review here the prion-like behavior of oncogenic p53 mutants that provides an explanation for their dominant-negative and gain-of-function properties and for the high metastatic potential of cancers bearing p53 mutations. The inhibition of the aggregation of p53 into oligomeric and fibrillar amyloids appears to be a promising target for therapeutic intervention in malignant tumor diseases.
Keywords: :
Introduction
The p53 tumor suppressor protein was discovered in 1979 by David Lane and coworkersCitation1 and later named the “Guardian of the Genome.” This designation is quite appropriate, since p53 is mutated in more than 50% of malignant tumors and it is suspected that the p53 pathway is affected in cases where no mutation is detected. The mutations of p53 lead not only to p53 loss-of-function, but also to negative-dominant effects (ND) (neutralization of the non-mutated p53) and gain-of-function (GF) effects. Recently, it has been proposed that the ND and GF effects occur via the oligomerization and aggregation of mutant p53 with wild-type p53 and other proteins in the p53 pathway, such as p63 and p73.
Prion diseases are rare maladies that are characterized by features that are also typical of many neurodegenerative diseases. Recent studies have suggested that the prion-related pathology is more general than previously thought and may encompass several neurodegenerative diseases.Citation2 Key proteins involved in these diseases such as Aβ, tau, α-synuclein, SOD1 and TDP43 can act as prions or prionoids; in addition, the pathological conformation of these proteins is transmitted both in animal and mammalian cell cultures in some cases.Citation3,Citation4 Intriguing new data on the aggregation of p53 have been produced recently. The ability of p53 to form aggregates and the kinetics associated with this process have been widely explored and discussed by different investigators.Citation5-Citation12 In addition, the prionoid behavior of p53 has been described by our groupCitation8 and by Forget et al.Citation13 These studies have enabled new opportunities for the investigation of the modulation of p53 and suggested new potential chemotherapeutic targets. However, the following questions remain to be answered: to what degree is p53 prionoid behavior important in cancer pathogenesis? Is cancer a prionoid disease? How can a prionoid behavior commonly found in degenerative diseases be relevant to a proliferative disease?
We review the amyloid and prion-like properties of p53 mutant and the similarities between this mutant and the mammalian prion protein (PrP).
Prion-Like Mechanisms and Prionoid Proteins
In 1982, Prusiner stated that “prions are small proteinaceous infectious particles that are resistant to inactivation by most procedures that modify nucleic acids”Citation14. The model used to explain the nature of the agent responsible for Transmissive Spongiform Encephalopathies (TSEs) was extremely controversial, as it stated the major protein PrP was infectious and could replicate in the absence of genomic nucleic acid.Citation15,Citation16 Since then, PrP has been characterized as a disease isoform known as PrP scrapie (PrPSc). This protein is partially resistant to proteinase K digestion and derived from a protease-sensitive endogenous protein known as cellular PrP (PrPC).Citation17,Citation18 The two isoforms have different conformations: PrPC is rich in α-helices, whereas PrPSc has numerous β-sheets; these differences confer different physicochemical properties to these proteins.Citation19,Citation20
The inheritance and transmissibility of PrPSc are programmed into the conformation of the protein, as PrPSc acts as a template that promotes PrPC misfolding and the formation of more PrPSc21–23. PrPSc is an insoluble protein that forms unorderedCitation24 and ordered aggregates.Citation25 For this reason, the determination of its three-dimensional structure is still a challenge. The mechanism behind prion conversion and aggregation is thought to involve protein self-replication as first described by GriffithCitation26 and later demonstrated by different authors.Citation21-Citation23 Ordered PrP polymers with intermolecular β-sheet structures form amyloid fibrils, most likely through a seeding-nucleation mechanism in which the rate-limiting factor for fibrillization is the formation of a nucleus.Citation27 This kinetic barrier arises from thermodynamically unfavorable self-association steps that build oligomers of different sizes and generate ordered nuclei. This nucleation step (or lag phase) produces more seeds that attract monomers once the nuclei are formed, resulting in a phase of rapid growth.Citation27 In the past 10 y, it has been found that polyanions such as RNA, DNA and glycosaminoglycans can participate as cofactors in the prion conversion process.Citation28-Citation31
The seeding-nucleation model is not exclusive to PrP fibrillization. A similar model is used to describe the process of crystallization,Citation32 the polymerization of actin,Citation33 the assembly of microtubulesCitation34 and the aggregation of amyloid and other proteins. Amyloid formation is known to cause other diseases characterized by similar misfolding processes. In each of these diseases, a major protein forms intra- or extracellular aggregates, leading to cellular dysfunctionCitation35; in particular, Aβ and Tau protein are involved in Alzheimer disease (AD), α-synuclein is involved in Parkinson disease (PD), huntingtin is involved in Huntington’s disease and superoxide dismutase (SOD) and TDP-43 are involved in amyothropic lateral sclerosis (ALS).
Over the last decade, many authors have shown that altered forms of other amyloid proteins are able to convert the normal form of the protein into misfolded proteins. These proteins have been called “prionoids” or “prion-like” proteins. The in vitro seeding of Aβ fibrils results in aggregates with the same morphology as that of the seedCitation36; a similar phenomenon has been observed for prion strains. The intracerebral injection of brain homogenates derived from AD patients induces disease in transgenic mice and rats expressing the amyloid protein (Aβ precursor).Citation37,Citation38 Mutant forms of SOD1 and TDP-43 are also able to seed misfolding of the wild-type protein both in vitro and in cell cultures.Citation4,Citation39
Self-propagation and template misfolding are basic characteristics of a prion; however, the ability to replicate in a new host is another defining characteristic of the prion. All proteins that produce nucleation and template misfolding are potentially transmissible in cells or organisms. Proteins that form extracellular aggregates (such as those formed by the Aβ peptide) can be seeded outside the cell; however, intracellular aggregates (such as those formed by the Tau protein) appear to involve more complex mechanisms of transcellular propagation. The observation of Lewy bodies (accumulations of α-synuclein in dopaminergic neurons) in fetal mesencephalic neurons transplanted into Parkinson disease patients provided early evidence that intracellular proteins can be misfolded through cell-to-cell transmission.Citation3 Intracerebral injections of Tau aggregates into mice also induced the aggregation of native Tau, similarly suggesting that transmission between cells could occur.Citation40 Whereas the mechanisms behind cell-to-cell transmission are still being investigated, some light has been shed on the active release of fibrilsCitation41 from one cell into the media, the release of these fibrils into recipient cellsCitation42 and their direct contact with other cells or their involvement with exosomes.Citation43 Laboratory experiments demonstrated the host-to-host transmission of AA amyloidosis.Citation44 However, no epidemiological data support the natural host-to-host transmission of any of these diseases other than TSEs.
It is unlikely that prion-like properties are exclusively characteristic of proteins involved in neurodegenerative diseases. The prionoid behavior of mutant p53 (i.e., the ability to aggregate) could be related to other cellular processes such as proliferation in cancer cells.Citation7-Citation9,Citation45 Bacterial surface proteins can act as prionoids.Citation46 The prion-related protein cytoplasmic polyadenylation element-binding (CPEB) of sensory neurons of Aplysia forms prion-like multimers, leading to long-term improvements in memory.Citation47
Transmissible proteins are found in both pathological and normal processes. Because all proteins can aggregate and form amyloid structures under certain circumstances (a general property of polypeptide chains),Citation48 prion-like properties may be more widespread than currently believed and self-propagation could be an evolutionarily relevant property aimed at spreading a specific effect.
p53 Aggregation and its Prion-Like Effect
The p53 protein is subject to several post-translational modifications and interacts with numerous other proteins. p53 is active in the form of a homotetramer of 393 amino acid residues. This protein is involved in several processes in the cell, including cell cycle arrest and apoptosis. Indeed, the flexible structure of p53 allows it to undergo different post-translational modifications in response to stress-related signals such as DNA damage. Each chain is composed of different domains: the N-terminal region containing the transactivation domain and a proline-rich segment; the central core domain (also known as the DNA-binding domain); and the C-terminal region, which includes the tetramerization domain and a natively unfolded regulatory domain.Citation49
The presence of mutations in the central core domain is very commonly encountered in cancer (affecting more than 50% of all cases), and a number of hot-spots have been described. Even though some of these mutations involve only a single amino acid, they can cause important loss-of-function or gain-of-function effects. The loss of function of WT p53 can disrupt many cellular processes and may induce the overexpression of genes previously repressed by p53. This appears to be the case for the ABCB1 gene (previously known as MDR1), which is involved in multidrug resistance.Citation50 Gain-of-function effects may lead to the induction of the expression of genes unaffected by the wild-type form of the protein either through the binding of p53 to non-canonical sequences of DNA or through the interaction of p53 with distinct transcription factors and the consequent enhancement of their activity (for excellent reviews, see refs. Citation51 and Citation52). Mutant p53 tends to be associated with a dominant-negative effect.Citation53 When one of the alleles of the gene is mutated and the other remains in its native form, the wild-type functions of p53 are abolished. The most traditional explanation for this observation involves the formation of heterotetramers containing both mutant and wild-type forms of the protein that are inactive.Citation54 Recent data suggest that this effect could be associated with the misfolding and aggregation of mutant p53 and the resultant prion-like effect on the wild-type form of the proteinCitation8,Citation9,Citation13,Citation55 (see ). One of the explanations offered in the literature for the aggregation of p53 involves the exposition of aggregation-prone sequences in mutant p53 that are usually hidden within the hydrophobic motifs of the wild-type protein.Citation9 The p53 homologous proteins p63 and p73 have similar aggregating-prone sequences () that enable their co-aggregation with p53, most likely through the central core domain.Citation9 This co-aggregation occurs despite the existence of only 38% homology in the oligomerization domain of these proteins.Citation56 Nevertheless, the interaction between mutant p53 and p63 has been shown to be related to important gain-of-function effects in cancer that appear to be the result of either the enhanced expression of genes targeted by p63Citation57,Citation58 or the binding of p63-p53 to unusual DNA sequences rather than to the loss of p63 DNA binding ability.Citation59 The existence of multiple isoforms of these three proteins further complicates these interactions and their effects, and the interactions between these proteins may lead to different cellular fates.Citation52
Figure 1. p53 prionoid aggregation scheme. (1) WTp53 suffers dominant-negative effect from mutant p53, acquiring a misfolded conformation, which leads to aggregation. (2) MDM2 and HSP70 might stabilize mutant p53 by forming structures named “pseudo-aggregates,” which can be involved in p53 intracellular aggregation.Citation71 (3) The co-aggregation of p53 with its partalogs, p63 and p73, has been described by Xu et al.Citation9 (4) p53 displays prionoid characteristics, since mutp53 aggregates are able to induce WTp53 conversion to the misfolded, aggregation-prone conformation.Citation8 These p53 aggregates might be captured by other cells through macropinocitosis.Citation13
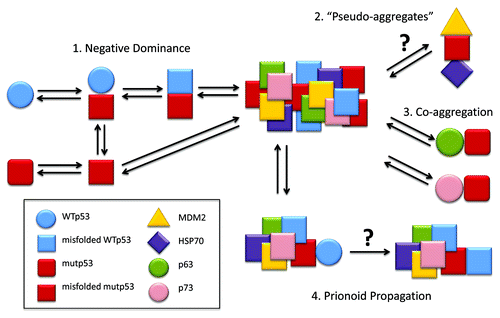
Table 1. Predicted aggregation behavior of cancer and neurodegenerative proteins
In Silico Analyses of Aggregation-Prone Sequences of p53, p63 and p73 and Comparison with Mammalian Prion Protein (PrP)
As mentioned previously, p53 is an unstable protein that forms amyloid aggregates in vitro and in vivo.Citation6,Citation8,Citation9,Citation12,Citation45,Citation60,Citation61 For this reason, we decided to evaluate the propensity of p53, p63 and p73 to form aggregates using prediction programs and comparing sequences of these proteins with sequences of other aggregation-prone protein sequences, including PrP. The prediction program TANGOCitation62 was used to identify hot-spots of p53 with a high propensity for β-aggregation; a sequence in its DNA binding domain that was experimentally shown to promote nucleate aggregation was identified.Citation9 The aggregation of p53 involves a self-propagation mechanism associated with a seed-nucleation model; an initial lag-phase characterized by the formation of p53 oligomers is followed by the exponential growth of amorphous and fibrillar aggregates.Citation6,Citation8 As is the case of prions and other prion-like diseases, these oligomers and aggregates are toxic to cells.Citation6,Citation8 The use of other predictive algorithms (see Citation63 for review) such as AGGRESCAN,Citation64 WALTZCitation65 and ZYGGREGATORCitation66 show that p53 has many hot-spots for aggregation (including amyloid sequences) that promote conversion to an amyloid state (; ). These tendencies are comparable to those of proteins involved in other prion-like diseases (). Interestingly, p63 and p73 display as high of a propensity to form aggregates as p53.
Figure 2. Human p53 primary sequence and aggregation hot spots colored according to the bioinformatics prediction method used. Aggrescan (red), Tango (light green), Aggrescan and Tango (brown), Aggrescan and Waltz (purple).

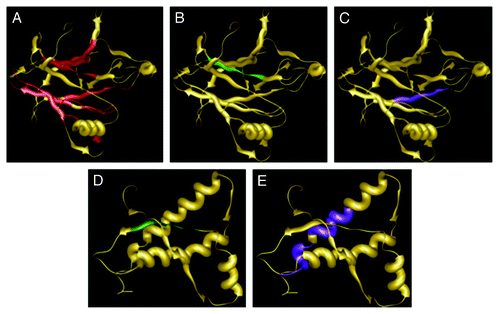
When only the core domain of p53 (p53C) is analyzed with these programs, significant aggregating segments of the protein are found to occur close to sequences where hot-spot mutations are found (). Also, p53 appears to form aggregates more easily than the murine PrP globular domain ().
Figure 3. Solution structure of human p53 DNA binding domain (PDB: 2FEJ) (A-C) and mouse PrP C-terminal domain (PDB: 1AG2) (DandE) with aggregation hot spots predicted by Aggrescan (A), Tango (BandD), and Waltz (C and E). AGGRESCAN hot spots predicted for mouse PrP are located at the N-terminal domain not represented by this model.
The discovery that unstructured p53 mutants drive changes in conformation in wild-type p53Citation67 demonstrated the template misfolding property and the seeding property of p53.Citation7-Citation9 The in vitro transmissibility of p53 was also recently demonstrated. Wild-type p53 aggregates were shown to enter HeLa and NIH3T3 cells (most likely via macropinocytosis) and induce intracellular p53 aggregation.Citation13
In their review article, Antony et al.Citation68 emphasized the likely role of protein-only inheritance and prions in cancer. Genetic studies with yeast showing that prions confer dominant phenotypes with cytoplasmic inheritance to these organisms indicate that several of these proteins have mammalian functional homologs.
Molecular Partners Involved in p53 Aggregation
Certain molecular partners of p53 could directly interfere with the propensity of this protein to form intracellular aggregates and thereby affect the normal cellular trafficking of the protein. In addition, the co-aggregation of p53 and many cellular proteins appears to occur in different cellular processes.Citation69,Citation70 Certain interactions have been proposed to stabilize p53 mutants in cells. The transient but recurrent interaction between p53 and the cellular chaperone HSP70 (Heat-Shock Protein 70) may promote an increase in the half-life of mutant p53 protein; in addition, in the presence of MDM2, these “pseudoaggregates” can form stable amyloid-like structuresCitation71 that appear to be associated with an altered structural conformation of p53.
Kirilyuk et al.Citation72 identified a highly disordered region of the acetyltransferase p300 with characteristics similar to those of prion-like domains. This region provides an interface with which misfolded proteins (including p53) can interact and aggregate. These findings demonstrate that p53 aggregates are generally heterogeneous.
In addition to considering the interaction between p53 and different proteins that cause aggregation and the formation of amyloid-like pseudo-aggregates, we must also consider the possible involvement of other molecules such as lipids and polyanions in these processes. Cholesterol secosterol aldehydes, which are lipids related to chronic inflammation, have been shown to promote p53 aggregation, with an amyloid nature.Citation73 In addition, evidence suggests that the interaction of p53 with a consensus DNA sequence stabilizes this protein and inhibits its aggregationCitation74; however, it is also possible that longer DNA chains could lead to polymerization.Citation75 Although the interaction between p53 and RNA has been reported,Citation76-Citation78 aggregates produced by this interaction have not yet been identified. Poly-adenylated RNA has been found in aggresomes that contain several key proteins (including p53) involved in cancer and neurodegenerative diseases.Citation69 Zanzoni et al.Citation79 claim that the interaction between p53 and its own mRNA might limit the amount of protein produced and thereby regulate p53 expression and aggregation. Finally, the mode of entrance of p53 prionoid aggregates into cellsCitation13 (i.e., via macropinocytosis) suggests that p53 may also interact with glycosaminoglycans (GAGs), similar to the interaction of PrP with GAGs.Citation31 In addition, certain molecular partners of p53 could directly interfere with its propensity to form intracellular aggregates and thereby affect the normal cellular trafficking of the protein.
More recently, the AB domain of the tumor suppressor retinoblastoma (RB) was shown to have aggregation properties similar to those of the p53 tumor suppressorCitation80; this finding is particularly relevant, as both cellular regulators are found inactivated in many cancers.
The Effect of p53 Mutation and Aggregation on Protein Trafficking
Extensive studies indicate that p53 acts as a major traffic controller in the cellular signaling network. In tumors, the main controllers of many cell signaling pathways (such as p53) are disrupted. Consequently, these cells acquire the ability to replicate in perpetuity, thus favoring the progression of cancer.Citation81 The p53 network is only activated when cells are damaged. This protein shuts down the proliferation of stressed cells by inhibiting progress through the cell cycle. In many cases, it can even trigger apoptosis to contain DNA damage and protect the organism.Citation82
The localization of p53 in the nucleus plays an important role in its response, as the regulation of transcription is one of the key functions of p53. The active transport of p53 toward the nucleus by dynein and the microtubule network has been described previouslyCitation83 and specific nuclear localization signals in the C-terminus of p53 contribute to this process. In the nucleus, regulatory mechanisms control the export of p53 to the cytosol. The p53 protein contains two nuclear export sequences: one in the C-terminal oligomerization domainCitation84 and one in the N-terminal MDM2 binding region.Citation85 The ability of p53 to be exported is enhanced by the action of MDM2,Citation84 although export is not absolutely dependent on the presence of this protein.Citation85,Citation86
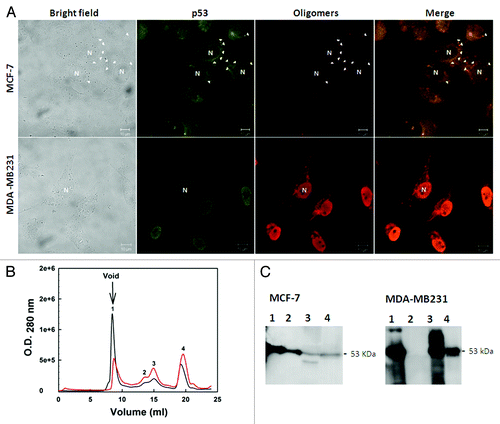
Previous studies have described the formation of amyloid-like aggregates during oligomerization. The formation of these aggregates is driven by the transactivation domains of p53,Citation61,Citation70,Citation87 which could affect the subcellular localization of the protein. Several cancers are characterized by an abnormal accumulation of wild-type or mutant p53 in the cytoplasm or in the nucleus of the cell.Citation88 We described a massive accumulation of p53 amyloid oligomers in MDA-MB-231 (mutant R280K p53) breast cancer cell nucleiCitation8 ().
Figure 4. Detection of native and aggregated p53 in breast cancer cell lines. (A) MCF-7 (wild-type p53) and MDA-MB 231 (mutated p53) cells were labeled with anti-p53 (DO-1) and anti-oligomer (A11) primary antibodies. The first column shows the bright field images, the second column shows p53-labeling, the third column shows the labeling of aggregates, and the last column shows the merged images of p53 labeling and aggregate labeling. The images were obtained at 63 000 magnification. (B) size exclusion chromatography fractions (SEC) of the extract of the MCF-7 (red line) and MDA-MB 231 (black line) tumoral cell lines. Western blotting against p53 was performed for the eluted fractions. (C) Aggregated p53 eluted in the column void volume. Extracted from ref. Citation8.
Therapeutic Approaches to Preventing Aggregation
The self-template-associated propagation of a protein with an altered conformation causes pathologies with characteristics of prion-like diseases. To understand this propagation process, it is important to manipulate the course of disease.
Recent suggestions that these diseases can also be transmitted from cell to cell offer the possibility of new therapeutic strategies involving the inhibition of the secretion and uptake of the pathogenic forms of these proteins.
Some treatment strategies aim to decrease protein production. RNA interference (RNAi)-based systems modulate gene expression at both transcriptional and posttranslational levels.Citation89 This latter method represents a very interesting strategy because it is highly specific (a desired characteristic in pharmacological drugs).Citation89 Such a strategy could be adopted for targets involving diseases caused by dominant gain-of-function mutations where the normal variant must be distinguished from the mutated form and where concentrations of the mutated protein in the cell must be decreased. This strategy has been tested with different diseases including ALS, HA, AD, PD and cancer.Citation89 The reactivation of p53 wild-type function (i.e., the induction of apoptosis) was performed with synthetic small inhibiting RNAs against mutant p53.Citation90 Decreasing the concentrations of mutant protein may decrease the aggregation of WT p53 and the co-aggregation of p53 with other suppressor proteins. Although this strategy shows promise, the delivery of RNAi reagents into cells and the stability of these compounds represent major obstacles.
Increasing intracellular and extracellular clearance also represents a method of eliminating toxic protein aggregates. Proteasomes are cellular components that perform autophagy and are responsible for proteolysis in eukaryotic cells. These cellular structures ensure proteostasis and control the quality of the cytoplasm: deficient proteasomes can promote protein aggregation by degrading damaged, misfolded and mutant proteins, whereas autophagy eliminates protein aggregates.Citation91 The regulation of autophagy may be therapeutically interesting. The upregulation of autophagy limits the accumulation of mutant Huntington and mutant α-synuclein.Citation92 The upregulation of autophagy has been shown to prevent cancer, but it can also lead to tumor survival in the later stages of tumorigenesis.Citation92 The main obstacle to the development of these strategies is the lack of known activators of this cellular machinery. The degradation of internalized misfolded proteins by autophagy and the inhibition of cellular uptake of these proteins could block disease progression.
Recently, we showed that resveratrol stabilizes wild-type p53 in breast cancer cell lines (MCF-7) and promotes apoptosis.Citation93 The transient transfection of the wild-type p53-GFP gene renders H1299 (originally lacking p53) more sensitive to resveratrol and more responsive to its pro-apoptotic properties. We propose that the combined use of resveratrol and other means of keeping active p53 cells (such as gene therapy using wild-type p53 gene or chemicals to rescue the function of p53) can be used therapeutically in cancer.Citation93
Immunotherapies for amyloid proteins have been tested with promising results, leading to the development of vaccine-based clinical trials. Antibodies can disaggregate proteins, promote their extracellular degradation and block their entry into cells.Citation94
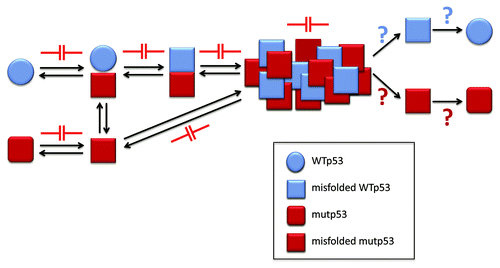
The prevention of aggregation represents a strategy that has been more widely studied. It is believed that misfolded proteins and/or small oligomers (intermediates of the aggregation process) are among the most toxic species that trigger disease. This process leads to the final formation of large amorphous aggregates or fibrils that are toxic in some studies and non-toxic in other studies.Citation48 This strategy is therefore currently focused on identifying molecules that interfere with the generation of aggregation nuclei and with the accumulation of misfolded proteins and pre-fibrillar aggregates. Natural or synthetic molecules that stabilize proteins/peptides, inhibit oligomerization and/or inhibit fibrillization have been studied. sketches the potential therapeutic targets in p53 aggregation.
Figure 5. p53 aggregation inhibition as a new approach for cancer chemotherapy. The aggregation process of p53 might be blocked in the many steps involved, including p53 misfolding and the prionoid effect. Also, as for other misfolding diseases, the oligomeric and aggregated forms might be destroyed, releasing both mutant and WT p53, whose destiny could include stabilization, with the recovery of transcriptional activity, or increased degradation, which could liberate the cell from the mutant p53 form and its deleterious effects.
p53 therapeutic approaches based on wild-type p53
Several anticancer therapies are based on the use of wild-type p53 or the targeting of p53-dependent pathways to induce cell death.Citation95,Citation96 Numerous studies have described the development of new strategies to facilitate the persistence of active p53 in cells and the promotion of its ability to function as a tumor suppressor.
Although p53 appears to have therapeutic potential, because it is not a receptor or an enzyme (the most conventionally studied targets for drug development), it was considered a poor drug target for a long time.Citation97 However, advances in drug design and scientific knowledge on p53 accumulated over the course of more than 30 y have enabled the development of new therapies based on restoring p53 function that have been applied in preclinical and clinical studies.Citation98
The main strategies used to restore pathways controlled by p53 include gene therapy, the modulation of post-translational modifications of p53 and the inhibition of the interaction between p53 and MDM2.
The insertion of wild-type p53 in these tumor cells has been attempted in several types of tumors that express mutant forms of p53 or are unable to express the protein and are therefore more resistant to conventional chemotherapy. Gene therapy with a p53 adenovirus vector produced antitumor efficacy in phase I, II and III clinical trials.Citation97 This type of therapy might in theory be useful for p53-null individuals; however, inserting a new source of WT p53 in cells without shutting down the endogenous mutant p53 expression might lead to an increase in aggregation.
Other approaches involving the use of tenovin-1 and tenovin-6 were effective in stabilizing cellular levels of p53. These compounds inhibit the deacetylase activity of SIRT1 and SIRT2 enzymes and promote the maintenance of stable levels of acetylated p53. Acetylation limits the degradation of p53 by MDM2 and is therefore crucial for its apoptotic function. The inhibition of the nuclear export of p53, which stimulates apoptosis and inhibits tumor growth, has also been described as a strategy for activating p53.Citation97 Other compounds act by blocking the interaction between p53 and MDM2, thus reducing the degradation of p53.Citation99 Nutlins, a class of MDM2 antagonists, and RITA (reactivation of p53 and induction of tumor cell apoptosis), a drug that interacts directly with wild-type p53 by preventing MDM2-mediated degradation, are two examples of such compounds. This strategy can also involve silencing the expression of MDM2 via transcriptional inhibitors that bind peptides and thereby impair p53-MDM2 interaction and increase the expression of ARF, a protein that inhibits MDM2.Citation100,Citation101
Mutant p53
The ability of mutant p53 to generate gain-of-function effects imply the necessity of interfering directly with this protein. The aggregation or accumulation of mutant p53 in cancer cells represents a property to be exploited (). Various drugs can destabilize mutant p53 (thus abolishing its acquired functions), stabilize mutant p53 (to recover, at least partially, the wild-type p53 functions that lead to apoptosis), stimulate p53 degradation pathways (to eliminate the noxious protein from the cell) and, theoretically, inhibit the prionoid properties of mutant p53 to avoid the spreading of this effect. When mutant p53 is inactivated (loss-of-function mutations), it can be reactivated to exert wild-type p53 functions ().
PRIMA-1, a drug that rescues the function of mutated p53, is able to form adducts with thiol groups of the mutated p53 core domain.Citation102 These adducts reactivate the mutated form of the protein and promote apoptosis in tumor cells. Another drug (MIRA-1) also appears to exert properties similar to those of PRIMA-1 and is more effective at inducing cell death. This drug appears to rescue mutant p53 function by shifting the equilibrium between native/denatured p53 forms and native conformation/active proteins.Citation103
The disruption of mutant p53 function could also be achieved by preventing its interaction with other transcription factors. The small-molecule RETRA has been shown to inhibit the interaction between mutant p53 and p73, thus restoring p73 function.Citation104 In addition, a number of peptides can promote the reactivation of wild-type function in mutant p53. Peptides corresponding to parts of the C-terminus domain of p53 have been suggested to restore the ability of mutant p53 to induce apoptosis.Citation105,Citation106
Recent works have described novel p53 active compounds that interfere with p53 aggregation. Rational drug design was used to analyze the aggregation of the p53 mutant Y220C using different compounds that bind to a small cavity in the C-terminal domain of this protein.Citation10 Another study of this mutant described the effects of a compound directed at the Y220C mutant that could reactivate properties of p53 and restore both transcriptional and non-transcriptional p53 functions.Citation107
Conclusions
The aggregation of mutant p53 (amyloid oligomers and fibrils) confers a prion-like activity on the native protein, converting it into an inactive form. The detection of amyloid aggregates in biopsied breast cancer tissues (especially in aggressive tumors) demonstrates the relevance of this prionoid behavior in cancer pathogenesis.Citation8,Citation45
The aggregation of mutant p53 induces the aggregation of WT p53, p63 and p73.Citation9 Recent findings showing that aggregates of p53C could be internalized by cells and co-aggregate with endogenous p53 protein corroborate the prionoid character of p53 aggregates.Citation13 The prion-like behavior of oncogenic p53 mutants ( and ) explains the dominant-negative and gain of function properties of these mutants and the high metastatic potential of cancers bearing p53 mutations. All these recent data have opened several questions concerning the relevance of aggregates in propagating the phenotype of “inactivated p53” (negative dominance) and altered function of other suppression proteins (such as p63 and p73) and other transcription factors (gain of function). There is strong evidence of intracellular prion-like propagation of misfolded p53, but the propagation among cells needs still to be proved, although it was demonstrated that aggregates of p53 can penetrate cell and induce aggregation.Citation13
The inhibition of the aggregation of p53 into oligomeric and fibrillar amyloids appears to represent a promising target for therapeutic intervention in cancer. Small molecules, small peptides and nucleic acid aptamers represent good candidates for this strategy.
| Abbreviations: | ||
| AD | = | Alzheimer disease |
| CPEB | = | cytoplasmic polyadenylation element-binding |
| HSP70 | = | heat-shock protein 70 |
| mutp53 | = | mutant p53 |
| p53C | = | p53 central core domain |
| PD | = | Parkinson disease |
| PrP | = | prion protein |
| PrPSc | = | PrP scrapie |
| RB | = | tumor suppressor retinoblastoma |
| SOD | = | superoxide dismutase |
| TSEs | = | transmissive spongiform encephalopathies |
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
Our laboratories were supported by grants from Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq awards and INCT Program), Fundação Carlos Chagas Filho de Amparo à Pesquisa do Estado do Rio de Janeiro (FAPERJ), Ministério da Saúde (Decit Program), Fundação do Câncer, and Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES).
Related Research Data
References
- Lane DP, Crawford LV. T antigen is bound to a host protein in SV40-transformed cells. Nature 1979; 278:261 - 3; http://dx.doi.org/10.1038/278261a0; PMID: 218111
- Soto C. Prion hypothesis: the end of the controversy?. Trends Biochem Sci 2011; 36:151 - 8; http://dx.doi.org/10.1016/j.tibs.2010.11.001; PMID: 21130657
- Li JY, Englund E, Holton JL, Soulet D, Hagell P, Lees AJ, Lashley T, Quinn NP, Rehncrona S, Björklund A, et al. Lewy bodies in grafted neurons in subjects with Parkinson’s disease suggest host-to-graft disease propagation. Nat Med 2008; 14:501 - 3; http://dx.doi.org/10.1038/nm1746; PMID: 18391963
- Grad LI, Guest WC, Yanai A, Pokrishevsky E, O’Neill MA, Gibbs E, Semenchenko V, Yousefi M, Wishart DS, Plotkin SS, et al. Intermolecular transmission of superoxide dismutase 1 misfolding in living cells. Proc Natl Acad Sci U S A 2011; 108:16398 - 403; http://dx.doi.org/10.1073/pnas.1102645108; PMID: 21930926
- Ishimaru D, Maia LF, Maiolino LM, Quesado PA, Lopez PC, Almeida FC, Valente AP, Silva JL. Conversion of wild-type p53 core domain into a conformation that mimics a hot-spot mutant. J Mol Biol 2003; 333:443 - 51; http://dx.doi.org/10.1016/j.jmb.2003.08.026; PMID: 14529628
- Ishimaru D, Andrade LR, Teixeira LS, Quesado PA, Maiolino LM, Lopez PM, Cordeiro Y, Costa LT, Heckl WM, Weissmüller G, et al. Fibrillar aggregates of the tumor suppressor p53 core domain. Biochemistry 2003; 42:9022 - 7; http://dx.doi.org/10.1021/bi034218k; PMID: 12885235
- Silva JL, Vieira TC, Gomes MP, Bom AP, Lima LM, Freitas MS, Ishimaru D, Cordeiro Y, Foguel D. Ligand binding and hydration in protein misfolding: insights from studies of prion and p53 tumor suppressor proteins. Acc Chem Res 2010; 43:271 - 9; http://dx.doi.org/10.1021/ar900179t; PMID: 19817406
- Ano Bom AP, Rangel LP, Costa DC, de Oliveira GA, Sanches D, Braga CA, Gava LM, Ramos CH, Cepeda AO, Stumbo AC, et al. Mutant p53 aggregates into prion-like amyloid oligomers and fibrils: implications for cancer. J Biol Chem 2012; 287:28152 - 62; http://dx.doi.org/10.1074/jbc.M112.340638; PMID: 22715097
- Xu J, Reumers J, Couceiro JR, De Smet F, Gallardo R, Rudyak S, Cornelis A, Rozenski J, Zwolinska A, Marine JC, et al. Gain of function of mutant p53 by coaggregation with multiple tumor suppressors. Nat Chem Biol 2011; 7:285 - 95; http://dx.doi.org/10.1038/nchembio.546; PMID: 21445056
- Wilcken R, Wang G, Boeckler FM, Fersht AR. Kinetic mechanism of p53 oncogenic mutant aggregation and its inhibition. Proc Natl Acad Sci U S A 2012; 109:13584 - 9; http://dx.doi.org/10.1073/pnas.1211550109; PMID: 22869713
- Wang G, Fersht AR. First-order rate-determining aggregation mechanism of p53 and its implications. Proc Natl Acad Sci U S A 2012; 109:13590 - 5; http://dx.doi.org/10.1073/pnas.1211557109; PMID: 22869710
- Lasagna-Reeves CA, Clos AL, Castillo-Carranza D, Sengupta U, Guerrero-Muñoz M, Kelly B, Wagner R, Kayed R. Dual role of p53 amyloid formation in cancer; loss of function and gain of toxicity. Biochem Biophys Res Commun 2013; 430:963 - 8; http://dx.doi.org/10.1016/j.bbrc.2012.11.130; PMID: 23261448
- Forget KJ, Tremblay G, Roucou X. p53 Aggregates penetrate cells and induce the co-aggregation of intracellular p53. PLoS One 2013; 8:e69242; http://dx.doi.org/10.1371/journal.pone.0069242; PMID: 23844254
- Prusiner SB. Novel proteinaceous infectious particles cause scrapie. Science 1982; 216:136 - 44; http://dx.doi.org/10.1126/science.6801762; PMID: 6801762
- Bolton DC, McKinley MP, Prusiner SB. Identification of a protein that purifies with the scrapie prion. Science 1982; 218:1309 - 11; http://dx.doi.org/10.1126/science.6815801; PMID: 6815801
- McKinley MP, Bolton DC, Prusiner SB. A protease-resistant protein is a structural component of the scrapie prion. Cell 1983; 35:57 - 62; http://dx.doi.org/10.1016/0092-8674(83)90207-6; PMID: 6414721
- Basler K, Oesch B, Scott M, Westaway D, Wälchli M, Groth DF, McKinley MP, Prusiner SB, Weissmann C. Scrapie and cellular PrP isoforms are encoded by the same chromosomal gene. Cell 1986; 46:417 - 28; http://dx.doi.org/10.1016/0092-8674(86)90662-8; PMID: 2873895
- Chesebro B, Race R, Wehrly K, Nishio J, Bloom M, Lechner D, Bergstrom S, Robbins K, Mayer L, Keith JM, et al. Identification of scrapie prion protein-specific mRNA in scrapie-infected and uninfected brain. Nature 1985; 315:331 - 3; http://dx.doi.org/10.1038/315331a0; PMID: 3923361
- Riek R, Hornemann S, Wider G, Glockshuber R, Wüthrich K. NMR characterization of the full-length recombinant murine prion protein, mPrP(23-231). FEBS Lett 1997; 413:282 - 8; http://dx.doi.org/10.1016/S0014-5793(97)00920-4; PMID: 9280298
- Pan KM, Baldwin M, Nguyen J, Gasset M, Serban A, Groth D, Mehlhorn I, Huang Z, Fletterick RJ, Cohen FE, et al. Conversion of alpha-helices into beta-sheets features in the formation of the scrapie prion proteins. Proc Natl Acad Sci U S A 1993; 90:10962 - 6; http://dx.doi.org/10.1073/pnas.90.23.10962; PMID: 7902575
- Telling GC, Parchi P, DeArmond SJ, Cortelli P, Montagna P, Gabizon R, Mastrianni J, Lugaresi E, Gambetti P, Prusiner SB. Evidence for the conformation of the pathologic isoform of the prion protein enciphering and propagating prion diversity. Science 1996; 274:2079 - 82; http://dx.doi.org/10.1126/science.274.5295.2079; PMID: 8953038
- Kocisko DA, Come JH, Priola SA, Chesebro B, Raymond GJ, Lansbury PT, Caughey B. Cell-free formation of protease-resistant prion protein. Nature 1994; 370:471 - 4; http://dx.doi.org/10.1038/370471a0; PMID: 7913989
- Kocisko DA, Priola SA, Raymond GJ, Chesebro B, Lansbury PT Jr., Caughey B. Species specificity in the cell-free conversion of prion protein to protease-resistant forms: a model for the scrapie species barrier. Proc Natl Acad Sci U S A 1995; 92:3923 - 7; http://dx.doi.org/10.1073/pnas.92.9.3923; PMID: 7732006
- McKinley MP, Braunfeld MB, Bellinger CG, Prusiner SB. Molecular characteristics of prion rods purified from scrapie-infected hamster brains. J Infect Dis 1986; 154:110 - 20; http://dx.doi.org/10.1093/infdis/154.1.110; PMID: 2872252
- Merz PA, Somerville RA, Wisniewski HM, Manuelidis L, Manuelidis EE. Scrapie-associated fibrils in Creutzfeldt-Jakob disease. Nature 1983; 306:474 - 6; http://dx.doi.org/10.1038/306474a0; PMID: 6358899
- Griffith JS. Self-replication and scrapie. Nature 1967; 215:1043 - 4; http://dx.doi.org/10.1038/2151043a0; PMID: 4964084
- Jarrett JT, Lansbury PT Jr.. Seeding “one-dimensional crystallization” of amyloid: a pathogenic mechanism in Alzheimer’s disease and scrapie?. Cell 1993; 73:1055 - 8; http://dx.doi.org/10.1016/0092-8674(93)90635-4; PMID: 8513491
- Cordeiro Y, Machado F, Juliano L, Juliano MA, Brentani RR, Foguel D, Silva JL. DNA converts cellular prion protein into the beta-sheet conformation and inhibits prion peptide aggregation. J Biol Chem 2001; 276:49400 - 9; http://dx.doi.org/10.1074/jbc.M106707200; PMID: 11604397
- Supattapone S, Deleault NR, Rees JR. Amplification of purified prions in vitro. Methods Mol Biol 2008; 459:117 - 30; http://dx.doi.org/10.1007/978-1-59745-234-2_9; PMID: 18576152
- Silva JL, Lima LM, Foguel D, Cordeiro Y. Intriguing nucleic-acid-binding features of mammalian prion protein. Trends Biochem Sci 2008; 33:132 - 40; http://dx.doi.org/10.1016/j.tibs.2007.11.003; PMID: 18243708
- Vieira TC, Reynaldo DP, Gomes MP, Almeida MS, Cordeiro Y, Silva JL. Heparin binding by murine recombinant prion protein leads to transient aggregation and formation of RNA-resistant species. J Am Chem Soc 2011; 133:334 - 44; http://dx.doi.org/10.1021/ja106725p; PMID: 21142149
- Crespo R, Rocha FA, Damas AM, Martins PM. A generic crystallization-like model that describes the kinetics of amyloid fibril formation. J Biol Chem 2012; 287:30585 - 94; http://dx.doi.org/10.1074/jbc.M112.375345; PMID: 22767606
- Condeelis J. How is actin polymerization nucleated in vivo?. Trends Cell Biol 2001; 11:288 - 93; http://dx.doi.org/10.1016/S0962-8924(01)02008-6; PMID: 11413039
- Job D, Valiron O, Oakley B. Microtubule nucleation. Curr Opin Cell Biol 2003; 15:111 - 7; http://dx.doi.org/10.1016/S0955-0674(02)00003-0; PMID: 12517712
- Vendruscolo M, Knowles TP, Dobson CM. Protein solubility and protein homeostasis: a generic view of protein misfolding disorders. Cold Spring Harb Perspect Biol 2011; 3:a010454; http://dx.doi.org/10.1101/cshperspect.a010454; PMID: 21825020
- Petkova AT, Leapman RD, Guo Z, Yau WM, Mattson MP, Tycko R. Self-propagating, molecular-level polymorphism in Alzheimer’s beta-amyloid fibrils. Science 2005; 307:262 - 5; http://dx.doi.org/10.1126/science.1105850; PMID: 15653506
- Ma J, Wollmann R, Lindquist S. Neurotoxicity and neurodegeneration when PrP accumulates in the cytosol. Science 2002; 298:1781 - 5; http://dx.doi.org/10.1126/science.1073725; PMID: 12386337
- Rosen RF, Fritz JJ, Dooyema J, Cintron AF, Hamaguchi T, Lah JJ, LeVine H 3rd, Jucker M, Walker LC. Exogenous seeding of cerebral β-amyloid deposition in βAPP-transgenic rats. J Neurochem 2012; 120:660 - 6; http://dx.doi.org/10.1111/j.1471-4159.2011.07551.x; PMID: 22017494
- Furukawa Y, Kaneko K, Watanabe S, Yamanaka K, Nukina N. A seeding reaction recapitulates intracellular formation of Sarkosyl-insoluble transactivation response element (TAR) DNA-binding protein-43 inclusions. J Biol Chem 2011; 286:18664 - 72; http://dx.doi.org/10.1074/jbc.M111.231209; PMID: 21454603
- Clavaguera F, Bolmont T, Crowther RA, Abramowski D, Frank S, Probst A, Fraser G, Stalder AK, Beibel M, Staufenbiel M, et al. Transmission and spreading of tauopathy in transgenic mouse brain. Nat Cell Biol 2009; 11:909 - 13; http://dx.doi.org/10.1038/ncb1901; PMID: 19503072
- Plouffe V, Mohamed NV, Rivest-McGraw J, Bertrand J, Lauzon M, Leclerc N. Hyperphosphorylation and cleavage at D421 enhance tau secretion. PLoS One 2012; 7:e36873; http://dx.doi.org/10.1371/journal.pone.0036873; PMID: 22615831
- Kfoury N, Holmes BB, Jiang H, Holtzman DM, Diamond MI. Trans-cellular propagation of Tau aggregation by fibrillar species. J Biol Chem 2012; 287:19440 - 51; http://dx.doi.org/10.1074/jbc.M112.346072; PMID: 22461630
- Aguzzi A, Rajendran L. The transcellular spread of cytosolic amyloids, prions, and prionoids. Neuron 2009; 64:783 - 90; http://dx.doi.org/10.1016/j.neuron.2009.12.016; PMID: 20064386
- Zhang B, Une Y, Fu X, Yan J, Ge F, Yao J, Sawashita J, Mori M, Tomozawa H, Kametani F, et al. Fecal transmission of AA amyloidosis in the cheetah contributes to high incidence of disease. Proc Natl Acad Sci U S A 2008; 105:7263 - 8; http://dx.doi.org/10.1073/pnas.0800367105; PMID: 18474855
- Levy CB, Stumbo AC, Ano Bom AP, Portari EA, Cordeiro Y, Silva JL, De Moura-Gallo CV. Co-localization of mutant p53 and amyloid-like protein aggregates in breast tumors. Int J Biochem Cell Biol 2011; 43:60 - 4; http://dx.doi.org/10.1016/j.biocel.2010.10.017; PMID: 21056685
- Blanco LP, Evans ML, Smith DR, Badtke MP, Chapman MR. Diversity, biogenesis and function of microbial amyloids. Trends Microbiol 2012; 20:66 - 73; http://dx.doi.org/10.1016/j.tim.2011.11.005; PMID: 22197327
- Si K, Lindquist S, Kandel ER. A neuronal isoform of the aplysia CPEB has prion-like properties. Cell 2003; 115:879 - 91; http://dx.doi.org/10.1016/S0092-8674(03)01020-1; PMID: 14697205
- Stefani M, Dobson CM. Protein aggregation and aggregate toxicity: new insights into protein folding, misfolding diseases and biological evolution. J Mol Med (Berl) 2003; 81:678 - 99; http://dx.doi.org/10.1007/s00109-003-0464-5; PMID: 12942175
- Joerger AC, Fersht AR. Structural biology of the tumor suppressor p53. Annu Rev Biochem 2008; 77:557 - 82; http://dx.doi.org/10.1146/annurev.biochem.77.060806.091238; PMID: 18410249
- Chin KV, Ueda K, Pastan I, Gottesman MM. Modulation of activity of the promoter of the human MDR1 gene by Ras and p53. Science 1992; 255:459 - 62; http://dx.doi.org/10.1126/science.1346476; PMID: 1346476
- Freed-Pastor WA, Prives C. Mutant p53: one name, many proteins. Genes Dev 2012; 26:1268 - 86; http://dx.doi.org/10.1101/gad.190678.112; PMID: 22713868
- Muller PA, Vousden KH. p53 mutations in cancer. Nat Cell Biol 2013; 15:2 - 8; http://dx.doi.org/10.1038/ncb2641; PMID: 23263379
- Gualberto A, Aldape K, Kozakiewicz K, Tlsty TD. An oncogenic form of p53 confers a dominant, gain-of-function phenotype that disrupts spindle checkpoint control. Proc Natl Acad Sci U S A 1998; 95:5166 - 71; http://dx.doi.org/10.1073/pnas.95.9.5166; PMID: 9560247
- Chan WM, Siu WY, Lau A, Poon RY. How many mutant p53 molecules are needed to inactivate a tetramer?. Mol Cell Biol 2004; 24:3536 - 51; http://dx.doi.org/10.1128/MCB.24.8.3536-3551.2004; PMID: 15060172
- Silva JL, Rangel LP, Costa DC, Cordeiro Y, De Moura Gallo CV. Expanding the prion concept to cancer biology: dominant-negative effect of aggregates of mutant p53 tumour suppressor. Biosci Rep 2013; 33:e00054; http://dx.doi.org/10.1042/BSR20130065; PMID: 24003888
- Irwin MS, Kaelin WG Jr.. Role of the newer p53 family proteins in malignancy. Apoptosis 2001; 6:17 - 29; http://dx.doi.org/10.1023/A:1009663809458; PMID: 11321038
- Neilsen PM, Noll JE, Suetani RJ, Schulz RB, Al-Ejeh F, Evdokiou A, Lane DP, Callen DF. Mutant p53 uses p63 as a molecular chaperone to alter gene expression and induce a pro-invasive secretome. Oncotarget 2011; 2:1203 - 17; PMID: 22203497
- Martynova E, Pozzi S, Basile V, Dolfini D, Zambelli F, Imbriano C, Pavesi G, Mantovani R. Gain-of-function p53 mutants have widespread genomic locations partially overlapping with p63. Oncotarget 2012; 3:132 - 43; PMID: 22361592
- Strano S, Fontemaggi G, Costanzo A, Rizzo MG, Monti O, Baccarini A, Del Sal G, Levrero M, Sacchi A, Oren M, et al. Physical interaction with human tumor-derived p53 mutants inhibits p63 activities. J Biol Chem 2002; 277:18817 - 26; http://dx.doi.org/10.1074/jbc.M201405200; PMID: 11893750
- Lee AS, Galea C, DiGiammarino EL, Jun B, Murti G, Ribeiro RC, Zambetti G, Schultz CP, Kriwacki RW. Reversible amyloid formation by the p53 tetramerization domain and a cancer-associated mutant. J Mol Biol 2003; 327:699 - 709; http://dx.doi.org/10.1016/S0022-2836(03)00175-X; PMID: 12634062
- Rigacci S, Bucciantini M, Relini A, Pesce A, Gliozzi A, Berti A, Stefani M. The (1-63) region of the p53 transactivation domain aggregates in vitro into cytotoxic amyloid assemblies. Biophys J 2008; 94:3635 - 46; http://dx.doi.org/10.1529/biophysj.107.122283; PMID: 18199664
- Fernandez-Escamilla AM, Rousseau F, Schymkowitz J, Serrano L. Prediction of sequence-dependent and mutational effects on the aggregation of peptides and proteins. Nat Biotechnol 2004; 22:1302 - 6; http://dx.doi.org/10.1038/nbt1012; PMID: 15361882
- Hamodrakas SJ. Protein aggregation and amyloid fibril formation prediction software from primary sequence: towards controlling the formation of bacterial inclusion bodies. FEBS J 2011; 278:2428 - 35; http://dx.doi.org/10.1111/j.1742-4658.2011.08164.x; PMID: 21569208
- Conchillo-Solé O, de Groot NS, Avilés FX, Vendrell J, Daura X, Ventura S. AGGRESCAN: a server for the prediction and evaluation of “hot spots” of aggregation in polypeptides. BMC Bioinformatics 2007; 8:65; http://dx.doi.org/10.1186/1471-2105-8-65; PMID: 17324296
- Oliveberg M. Waltz, an exciting new move in amyloid prediction. Nat Methods. United States, 2010:187-8.
- Tartaglia GG, Vendruscolo M. The Zyggregator method for predicting protein aggregation propensities. Chem Soc Rev 2008; 37:1395 - 401; http://dx.doi.org/10.1039/b706784b; PMID: 18568165
- Milner J, Medcalf EA. Cotranslation of activated mutant p53 with wild type drives the wild-type p53 protein into the mutant conformation. Cell 1991; 65:765 - 74; http://dx.doi.org/10.1016/0092-8674(91)90384-B; PMID: 2040013
- Antony H, Wiegmans AP, Wei MQ, Chernoff YO, Khanna KK, Munn AL. Potential roles for prions and protein-only inheritance in cancer. Cancer Metastasis Rev 2012; 31:1 - 19; http://dx.doi.org/10.1007/s10555-011-9325-9; PMID: 22138778
- Latonen L, Moore HM, Bai B, Jäämaa S, Laiho M. Proteasome inhibitors induce nucleolar aggregation of proteasome target proteins and polyadenylated RNA by altering ubiquitin availability. Oncogene 2011; 30:790 - 805; http://dx.doi.org/10.1038/onc.2010.469; PMID: 20956947
- Lee SY, Jeong EK, Jeon HM, Kim CH, Kang HS. Implication of necrosis-linked p53 aggregation in acquired apoptotic resistance to 5-FU in MCF-7 multicellular tumour spheroids. Oncol Rep 2010; 24:73 - 9; PMID: 20514446
- Wiech M, Olszewski MB, Tracz-Gaszewska Z, Wawrzynow B, Zylicz M, Zylicz A. Molecular mechanism of mutant p53 stabilization: the role of HSP70 and MDM2. PLoS One 2012; 7:e51426; http://dx.doi.org/10.1371/journal.pone.0051426; PMID: 23251530
- Kirilyuk A, Shimoji M, Catania J, Sahu G, Pattabiraman N, Giordano A, Albanese C, Mocchetti I, Toretsky JA, Uversky VN, et al. An intrinsically disordered region of the acetyltransferase p300 with similarity to prion-like domains plays a role in aggregation. PLoS One 2012; 7:e48243; http://dx.doi.org/10.1371/journal.pone.0048243; PMID: 23133622
- Nieva J, Song BD, Rogel JK, Kujawara D, Altobel L 3rd, Izharrudin A, Boldt GE, Grover RK, Wentworth AD, Wentworth P Jr.. Cholesterol secosterol aldehydes induce amyloidogenesis and dysfunction of wild-type tumor protein p53. Chem Biol 2011; 18:920 - 7; http://dx.doi.org/10.1016/j.chembiol.2011.02.018; PMID: 21802012
- Ishimaru D, Ano Bom AP, Lima LM, Quesado PA, Oyama MF, de Moura Gallo CV, Cordeiro Y, Silva JL. Cognate DNA stabilizes the tumor suppressor p53 and prevents misfolding and aggregation. Biochemistry 2009; 48:6126 - 35; http://dx.doi.org/10.1021/bi9003028; PMID: 19505151
- Lyubchenko YL, Shlyakhtenko LS, Nagaich A, Appella E, Harrington RE, Lindsay SM. Polymerization of the dna binding fragment of p53 on dna: atomic force microscopy study. Scanning Microsc 1998; 12:455 - 63
- Riley KJ, James Maher L 3rd. Analysis of p53-RNA interactions in cultured human cells. Biochem Biophys Res Commun 2007; 363:381 - 7; http://dx.doi.org/10.1016/j.bbrc.2007.08.181; PMID: 17869221
- Samad A, Carroll RB. The tumor suppressor p53 is bound to RNA by a stable covalent linkage. Mol Cell Biol 1991; 11:1598 - 606; PMID: 1705009
- Yoshida Y, Izumi H, Torigoe T, Ishiguchi H, Yoshida T, Itoh H, Kohno K. Binding of RNA to p53 regulates its oligomerization and DNA-binding activity. Oncogene 2004; 23:4371 - 9; http://dx.doi.org/10.1038/sj.onc.1207583; PMID: 15064727
- Zanzoni A, Marchese D, Agostini F, Bolognesi B, Cirillo D, Botta-Orfila M, Livi CM, Rodriguez-Mulero S, Tartaglia GG. Principles of self-organization in biological pathways: a hypothesis on the autogenous association of alpha-synuclein. Nucleic Acids Res 2013; 41:9987 - 98; http://dx.doi.org/10.1093/nar/gkt794; PMID: 24003031
- Chemes LB, Noval MG, Sánchez IE, de Prat-Gay G. Folding of a cyclin box: linking multitarget binding to marginal stability, oligomerization, and aggregation of the retinoblastoma tumor suppressor AB pocket domain. J Biol Chem 2013; 288:18923 - 38; http://dx.doi.org/10.1074/jbc.M113.467316; PMID: 23632018
- Sebastian S, Azzariti A, Silvestris N, Porcelli L, Russo A, Paradiso A. p53 as the main traffic controller of the cell signaling network. [Landmark Ed] Front Biosci (Landmark Ed) 2010; 15:1172 - 90; http://dx.doi.org/10.2741/3669; PMID: 20515749
- Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature 2000; 408:307 - 10; http://dx.doi.org/10.1038/35042675; PMID: 11099028
- Giannakakou P, Sackett DL, Ward Y, Webster KR, Blagosklonny MV, Fojo T. p53 is associated with cellular microtubules and is transported to the nucleus by dynein. Nat Cell Biol 2000; 2:709 - 17; http://dx.doi.org/10.1038/35036335; PMID: 11025661
- Stommel JM, Marchenko ND, Jimenez GS, Moll UM, Hope TJ, Wahl GM. A leucine-rich nuclear export signal in the p53 tetramerization domain: regulation of subcellular localization and p53 activity by NES masking. EMBO J 1999; 18:1660 - 72; http://dx.doi.org/10.1093/emboj/18.6.1660; PMID: 10075936
- Zhang T, Prives C. Cyclin a-CDK phosphorylation regulates MDM2 protein interactions. J Biol Chem 2001; 276:29702 - 10; http://dx.doi.org/10.1074/jbc.M011326200; PMID: 11359766
- Freedman DA, Wu L, Levine AJ. Functions of the MDM2 oncoprotein. Cell Mol Life Sci 1999; 55:96 - 107; http://dx.doi.org/10.1007/s000180050273; PMID: 10065155
- Higashimoto Y, Asanomi Y, Takakusagi S, Lewis MS, Uosaki K, Durell SR, Anderson CW, Appella E, Sakaguchi K. Unfolding, aggregation, and amyloid formation by the tetramerization domain from mutant p53 associated with lung cancer. Biochemistry 2006; 45:1608 - 19; http://dx.doi.org/10.1021/bi051192j; PMID: 16460008
- Moll UM, Ostermeyer AG, Haladay R, Winkfield B, Frazier M, Zambetti G. Cytoplasmic sequestration of wild-type p53 protein impairs the G1 checkpoint after DNA damage. Mol Cell Biol 1996; 16:1126 - 37; PMID: 8622657
- Seyhan AA. RNAi: a potential new class of therapeutic for human genetic disease. Hum Genet 2011; 130:583 - 605; http://dx.doi.org/10.1007/s00439-011-0995-8; PMID: 21537948
- Martinez LA, Naguibneva I, Lehrmann H, Vervisch A, Tchénio T, Lozano G, Harel-Bellan A. Synthetic small inhibiting RNAs: efficient tools to inactivate oncogenic mutations and restore p53 pathways. Proc Natl Acad Sci U S A 2002; 99:14849 - 54; http://dx.doi.org/10.1073/pnas.222406899; PMID: 12403821
- Tanaka K, Matsuda N. Proteostasis and neurodegeneration: The roles of proteasomal degradation and autophagy. Biochim Biophys Acta 2014; 1843:197 - 204; http://dx.doi.org/10.1016/j.bbamcr.2013.03.012; PMID: 23523933
- Rubinsztein DC, Codogno P, Levine B. Autophagy modulation as a potential therapeutic target for diverse diseases. Nat Rev Drug Discov 2012; 11:709 - 30; http://dx.doi.org/10.1038/nrd3802; PMID: 22935804
- Ferraz da Costa DC, Casanova FA, Quarti J, Malheiros MS, Sanches D, Dos Santos PS, Fialho E, Silva JL. Transient transfection of a wild-type p53 gene triggers resveratrol-induced apoptosis in cancer cells. PLoS One 2012; 7:e48746; http://dx.doi.org/10.1371/journal.pone.0048746; PMID: 23152798
- Kaufman SK, Diamond MI. Prion-like propagation of protein aggregation and related therapeutic strategies. Neurotherapeutics 2013; 10:371 - 82; http://dx.doi.org/10.1007/s13311-013-0196-3; PMID: 23801258
- Lin Y, Benchimol S. Cytokines inhibit p53-mediated apoptosis but not p53-mediated G1 arrest. Mol Cell Biol 1995; 15:6045 - 54; PMID: 7565757
- Riou G, Barrois M, Prost S, Terrier MJ, Theodore C, Levine AJ. The p53 and mdm-2 genes in human testicular germ-cell tumors. Mol Carcinog 1995; 12:124 - 31; http://dx.doi.org/10.1002/mc.2940120303; PMID: 7893365
- Chen F, Wang W, El-Deiry WS. Current strategies to target p53 in cancer. Biochem Pharmacol 2010; 80:724 - 30; http://dx.doi.org/10.1016/j.bcp.2010.04.031; PMID: 20450892
- Brown CJ, Lain S, Verma CS, Fersht AR, Lane DP. Awakening guardian angels: drugging the p53 pathway. Nat Rev Cancer 2009; 9:862 - 73; http://dx.doi.org/10.1038/nrc2763; PMID: 19935675
- Chène P. Inhibiting the p53-MDM2 interaction: an important target for cancer therapy. Nat Rev Cancer 2003; 3:102 - 9; http://dx.doi.org/10.1038/nrc991; PMID: 12563309
- Issaeva N, Bozko P, Enge M, Protopopova M, Verhoef LG, Masucci M, Pramanik A, Selivanova G. Small molecule RITA binds to p53, blocks p53-HDM-2 interaction and activates p53 function in tumors. Nat Med 2004; 10:1321 - 8; http://dx.doi.org/10.1038/nm1146; PMID: 15558054
- Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, Kong N, Kammlott U, Lukacs C, Klein C, et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 2004; 303:844 - 8; http://dx.doi.org/10.1126/science.1092472; PMID: 14704432
- Lambert JM, Gorzov P, Veprintsev DB, Söderqvist M, Segerbäck D, Bergman J, Fersht AR, Hainaut P, Wiman KG, Bykov VJ. PRIMA-1 reactivates mutant p53 by covalent binding to the core domain. Cancer Cell 2009; 15:376 - 88; http://dx.doi.org/10.1016/j.ccr.2009.03.003; PMID: 19411067
- Bykov VJ, Issaeva N, Zache N, Shilov A, Hultcrantz M, Bergman J, Selivanova G, Wiman KG. Reactivation of mutant p53 and induction of apoptosis in human tumor cells by maleimide analogs. J Biol Chem 2005; 280:30384 - 91; http://dx.doi.org/10.1074/jbc.M501664200; PMID: 15998635
- Kravchenko JE, Ilyinskaya GV, Komarov PG, Agapova LS, Kochetkov DV, Strom E, Frolova EI, Kovriga I, Gudkov AV, Feinstein E, et al. Small-molecule RETRA suppresses mutant p53-bearing cancer cells through a p73-dependent salvage pathway. Proc Natl Acad Sci U S A 2008; 105:6302 - 7; http://dx.doi.org/10.1073/pnas.0802091105; PMID: 18424558
- Selivanova G, Iotsova V, Okan I, Fritsche M, Ström M, Groner B, Grafström RC, Wiman KG. Restoration of the growth suppression function of mutant p53 by a synthetic peptide derived from the p53 C-terminal domain. Nat Med 1997; 3:632 - 8; http://dx.doi.org/10.1038/nm0697-632; PMID: 9176489
- Kim AL, Raffo AJ, Brandt-Rauf PW, Pincus MR, Monaco R, Abarzua P, Fine RL. Conformational and molecular basis for induction of apoptosis by a p53 C-terminal peptide in human cancer cells. J Biol Chem 1999; 274:34924 - 31; http://dx.doi.org/10.1074/jbc.274.49.34924; PMID: 10574967
- Liu X, Wilcken R, Joerger AC, Chuckowree IS, Amin J, Spencer J, Fersht AR. Small molecule induced reactivation of mutant p53 in cancer cells. Nucleic Acids Res 2013; 41:6034 - 44; http://dx.doi.org/10.1093/nar/gkt305; PMID: 23630318