
Abstract
Prion diseases are caused by misfolding and aggregation of the prion protein (PrP). Pathogenic mutations such as Y218N and E196K are known to cause Gerstmann-Sträussler-Scheinker syndrome and Creutzfeldt-Jakob disease, respectively. Here we describe molecular dynamics simulations of these mutant proteins to better characterize the detailed conformational effects of these sequence substitutions. Our results indicate that the mutations disrupt the wild-type native PrPC structure and cause misfolding. Y218N reduced hydrophobic packing around the X-loop (residues 165–171), and E196K abolished an important wild-type salt bridge. While differences in the mutation site led PrP mutants to misfold along different pathways, we observed multiple traits of misfolding that were common to both mutants. Common traits of misfolding included: 1) detachment of the short helix (HA) from the PrP core; 2) exposure of side chain F198; and 3) formation of a nonnative strand at the N-terminus. The effect of the E196K mutation directly abolished the wild-type salt bridge E196-R156, which further destabilized the F198 hydrophobic pocket and HA. The Y218N mutation propagated its effect by increasing the HB-HC interhelical angle, which in turn disrupted the packing around F198. Furthermore, a nonnative contact formed between E221 and S132 on the S1-HA loop, which offered a direct mechanism for disrupting the hydrophobic packing between the S1-HA loop and HC. While there were common misfolding features shared between Y218N and E196K, the differences in the orientation of HB and HC and the X-loop conformation might provide a structural basis for identifying different prion strains.
Introduction
Prion diseases, or transmissible spongiform encephalopathies, are neurodegenerative diseases that include Creutzfeldt-Jakob disease (CJD), Gerstmann-Sträussler-Scheinker Syndrome (GSS) and fatal familial insomnia. Human prion diseases are extremely heterogeneous in terms of their clinical presentations and biomolecular characteristics.Citation1-Citation3 Different prion diseases can only be identified upon examination of the prion protein (PrP) aggregates in neuronal tissues.Citation4,Citation5 Despite differences in neuropathology, all prion diseases share the same causative agent, which is the scrapie form of the prion protein, PrPSc. The normal cellular prion protein (PrPC) is innocuous, but it may undergo misfolding and convert into PrPSc,Citation6,Citation7 which is toxic and infectious. Currently, the PrPSc conversion mechanism is not well understood, nor is there a clear understanding of how different types of prion diseases occur.
PrPC is expressed in neuronal cells as a membrane bound protein with two glycosylation sites. The N-terminus (residues 23–127) is highly flexible, but the C-terminal globular domain (residues 128–228) is structured in various species.Citation8-Citation11 The C-terminal globular domain consists of three helices (HA, HB, and HC) and two short β-strands (S1 and S2) (). At the C-terminus, there is a glycosylphosphatidylinositol (GPI)-anchor that tethers the PrPC to the cell membrane. Transgenic mice expressing full-length mouse PrP without the GPI anchor produces mostly unglycosylated PrP.Citation12,Citation13 In addition, these mice are susceptible to prion diseases.Citation12 This suggests that the protein-only portion of PrP (without glycosylation and the GPI anchor) is sufficient for PrPSc formation in mice. Proteinase K digestion of PrPSc aggregates in human patients indicate that residues ~90–230 are protected and thus these residues are considered to be the core of PrPSc.Citation14 The PrP fragment with residues 90–230 (PrP90–230) contains critical residues in the N-terminus that are required for PrPSc conversion.Citation15-Citation18 In vivo studies have also demonstrated that recombinant fibrils of PrP90–230 in mouse are infectious.Citation19 These findings suggest that PrP90–230 is the core of PrPSc that carries infectivity.
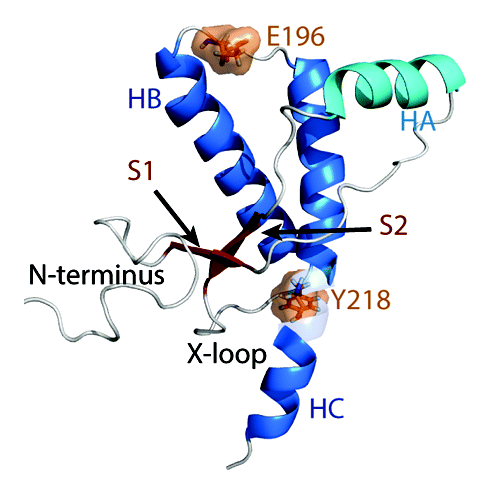
Figure 1. Native structure of the human PrP. Starting structure for WT PrP MD simulations. Structure of residues 128–228 was obtained from PDB 1QLX. Structure of the flexible N-terminus (residues 90–127), and C-terminus (residues 229–230) were constructed manually. Helices HB and HC are colored in blue and HA is colored in cyan. Native strands (S1 and S2) are colored in dark red. The remaining loop regions are in gray. Mutation sites are indicated in orange.
Nearly all (> 30) of the known single point pathogenic mutations in the human PrP are within residues 90–230.Citation20-Citation23 More than 20 of these single point pathogenic mutations are spread throughout the HB and HC regions. The lack of convergence into a hot spot of mutations makes it difficult to rationalize the mutation-induced misfolding pathway, as different mutations have different local effects on PrP dynamics. While several unfolding studies indicate that single point pathogenic mutations destabilize the PrPC structure, some mutations increase or have little effect on the thermodynamic stability of PrPC.Citation24,Citation25 Molecular dynamics (MD) studies of pathogenic mutations in the PrP hydrophobic coreCitation26 have identified misfolded structures in agreement with earlier MD studies of the human wild-type (WT) PrP using low pH to trigger misfolding.Citation27,Citation28
Disease-associated mutations Y218NCitation29 and E196KCitation30 cause GSS and familial CJD, respectively. These two mutants are different in terms of the amino acids mutated, and the mutation sites are separated by ~29 Å. Here, we used MD simulations to study the PrP dynamics of these two disease-associated mutants. We observed common misfolding events in agreement with previous MD studies of other pathogenic mutants.Citation26 However, certain structural features and the pathways of misfolding were different for Y218N and E196K. The structural differences provide us with a structure-based hypothesis regarding the formation of different prion strains.
Results
Major conformational change in the globular domain
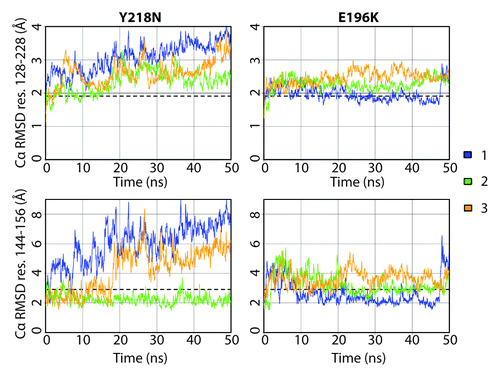
The Cα root-mean-square-deviation (RMSD) of the globular domain was used to monitor conformational changes in the structured region of PrP (). At the end of 50 ns, Y218N simulations 1 and 3 reached much higher Cα RMSDs (~3.5 Å) than that of the average from the WT simulations (1.9 Å). The Cα RMSD of simulation 2 at the end was about 2.5 Å, which was ~0.5 Å higher than that of the average WT simulations. Previous low pH MD simulations of human PrP have shown that the HA helix is highly mobile and can contribute to the high globular Cα RMSD.Citation31 To monitor the HA movement with respect to the core of the protein, the Cα RMSD of HA was measured () by using the stable core domain (residues 174–186 and 200–219) for alignment. The increasing Cα RMSD of HA in simulations 1 and 3 correlated with the increasing globular Cα RMSD. Simulation 2 had relatively low Cα RMSD of HA compared with that of the other simulations and the average WT Cα RMSD. Such Cα RMSD trend indicates that simulation 2 preserved the native HA orientation with respect to the core unlike the other two simulations of Y218N. The globular domain Cα RMSD of E196K was similar to that of the WT. The Cα RMSD of HA in E196K simulations stayed close to the average WT value, in contrast to Y218N. However, there was an increase in Cα RMSD of HA in E196K simulation 1 after 48 ns, which indicated a sudden HA movement with respect to the core.
Figure 2. Cα RMSD of the globular domain (residues 128–228) and HA (residues 144–156) for Y218N and E196K simulations (three simulations of each). The globular domain was aligned for calculating Cα RMSD residues 128–228, and the core domain (residues 174–186 and 200–219) was aligned for calculating Cα RMSD residues 144–156. Window averages (100 ps) of the Cα RMSD are shown for all mutant simulations. The average Cα RMSD value over the last 25 ns of the four WT simulations are shown as dashed lines.
HA detachment from core in mutant simulations
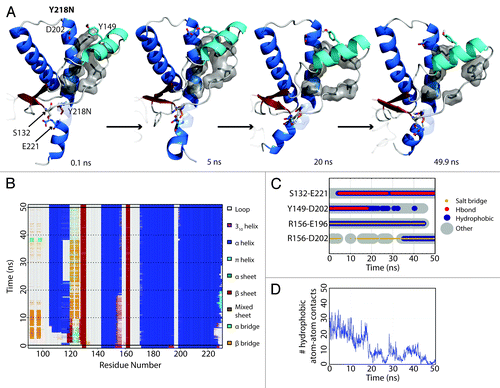
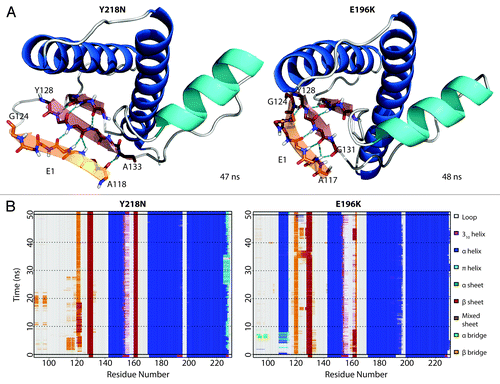
HA detached from the protein core in simulation 3 of Y218N (). The hydrophobic contacts between S1-HA loop and HC (42 atom contacts) in the starting structure were the main attractive force that held HA and HC together. Within the first nanosecond, D202 formed a hydrogen bond with Y149. At 5 ns, a rare hydrogen bond between S132 and E221 formed. This contact was only observed in WT simulations 0.57% of the time. At 18 ns, HA moved away from HC, which occurred ~13 ns after the formation of the S132-E221 nonnative hydrogen bond. The HA movement was characterized by three events that happened cooperatively: 1) a change from 310-helix to α-helix at the C-terminus of HA (); 2) the loss of a hydrogen bond between D202 and Y149 (); and 3) the loss of hydrophobic contacts between the S1-HA loop and HC (). HA remained in this nonnative position for the rest of the simulation.
Figure 3. HA detachment from the PrP core in Y218N simulation 3. (A) Structures taken at 0.1, 5, 20, and 40.9 ns of the simulation. Residues S132, Y149, D202 and E221 are shown as sticks. Residues shown in gray sticks with a transparent surface are P137, I139, F141 on the S1-HA loop and M205, V209, M213 on HC. (B) DSSP analysis on the protein secondary structure. (C) Contacts between pairs of listed residues over 50 ns in simulation. (D) Window average (100 ps) of the number of contacts between residues P137, I139, F141 on the S1-HA loop and M205, V209, M213 on HC.
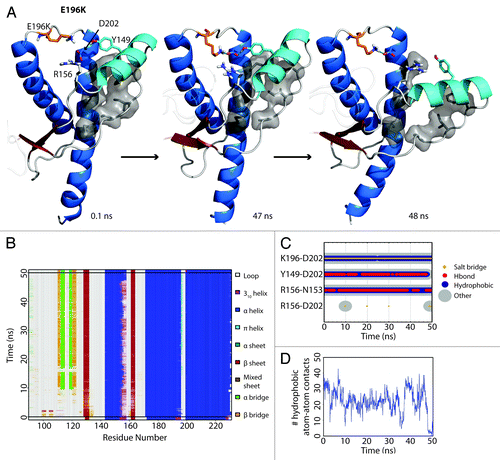
The starting structure of E196K had only minute differences from Y218N (Cα RMSD = 0.140), except that the mutant E196K had a new salt bridge between K196 and D202. The WT salt bridge between E196-R156 was abolished due to the E196K mutation. The hydrogen bond between D202 and Y149 formed within the first nanosecond of the simulation. After 48 ns in simulation 3, HA detached from HC () and was characterized by the three aforementioned events (). The nonnative salt bridge K196-D202 was particularly prominent in E196K simulation 3 and lasted throughout the course of the simulation (). Since the D202 side chain was occupied with K196, and the side chain of residue 196 became positively charged, R156 lost both of its WT salt bridge partners (residue 202 and 196). The salt bridge between D202 and R156 was occupied on average 1.84% of the time from 25–50 ns of the E196K simulations, unlike WT and Y218N (36% and 59% respectively). The remaining neighbors available for interacting with R156 were on HA. In particular, the side chain of N153 was available to interact with R156 ().
Figure 4. HA detachment from the PrP core in E196K simulation 1. (A) Structures taken at 0.1, 47, and 48 ns. Residues D202, Y149, R156 and K196 are shown as sticks. Residues shown in gray sticks with a transparent Van der Waals radius are P137, I139, F141 on the S1-HA loop and M205, V209, M213 on HC. (B) DSSP analysis on the protein secondary structure. (C) Contacts between pairs of listed residues over 50 ns in simulation. (D) Window average (100 ps) of the number of contacts between residues P137, I139, F141 on the S1-HA loop and M205, V209, M213 on HC.
Hydrophobic core packing and F198 solvent exposure
Both mutants had a more exposed hydrophobic core than that of WT, except for one simulation of E196K (). Exposure of the hydrophobic core was mainly attributed to HA movement, X-loop hydrophobic packing and also the solvent exposure of F198. The average solvent accessible surface area (SASA) of F198 in WT was only 34 Å2, while some of the mutant simulations achieved > 150 Å2 at some time points. F198 can be roughly classified into three conformational states by its solvent exposure: native state (SASA ~20 Å2) with F198 side chain buried between HB and HC; semi-exposed state (SASA ~80 Å2) with one side of the phenyl group exposed to solvent; and, fully exposed state (SASA ~140 Å2) with the side chain fully exposed to solvent. These conformations of F198 are illustrated in for WT, Y218N and E196K.
Figure 5. Solvent accessible surface area of the hydrophobic core and F198 in the Y218N and E196K simulations. The hydrophobic core was defined as residues 134, 137, 139, 141, 158, 161, 175, 176, 179, 180, 184, 198, 203, 205, 206, 209, 210, and 213–215. Window averages (100 ps) of the SASA are shown for all mutant simulations. The average SASA values over the last 25 ns of the WT simulations are shown in dashed lines.
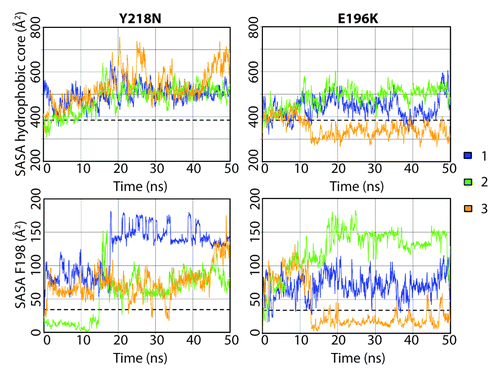
Figure 6. Salt bridge network around the F198 side chain. (A, B and C) Structures at specific time points of the WT simulation 2, Y218N simulation 1, and E196K simulation 2, respectively. Secondary structure color schemes are cyan for HA, blue for HB and HC, where the transparent helix is HC. Residues E/K196, F198, T199, D202, and R156 are shown in sticks and hydrogen bonds are indicated with dotted blue lines. (far right) Relevant contacts are plotted over time.
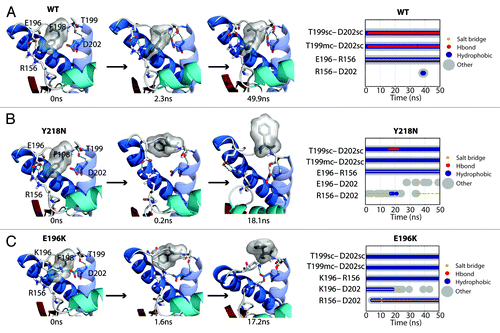
In WT simulations, F198 was mostly in its native state and occasionally visited the semi-exposed state. The backbone conformation of F198 was stabilized by its neighbor T199, which formed a capping box interactionCitation32 with D202 at the N-terminus of HC (). The WT native salt bridge between E196 and R156 also stabilized the backbone conformation of the HB-HC loop. This WT native salt bridge was preserved in Y218N (). However, for E196K, the mutation at residue 196 abolished the WT native salt bridge and directly triggered a mechanism that destabilized the conformation of F198 (). K196 and D202 formed a salt bridge in the starting structure. This new salt bridge caused the side chain of K196 to clash with F198. At 1.6 ns, the F198 side chain reached the semi-exposed state due to steric repulsion with K196. As R156 formed another salt bridge with D202 at 17.2ns, K196 was pushed out of the protein core and the side chain of F198 became fully exposed. The backbone conformation of the HB-HC loop was vulnerable at the very beginning, because K196 formed a salt bridge with D202, thereby preventing the formation of the capping box interaction between T199 and D202.
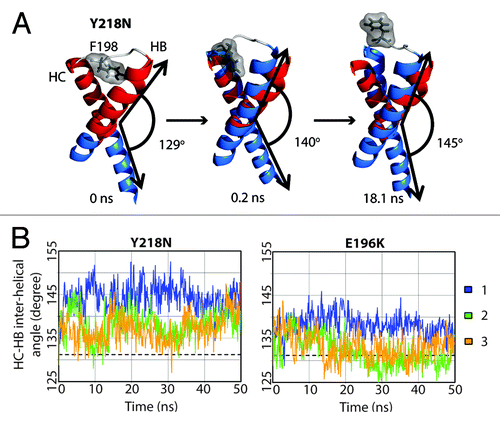
Y218N simulations also had a disrupted F198 hydrophobic pocket, but via an indirect mechanism, which involved an increase in the interhelical angle between HB and HC (). In general, Y218N had a larger interhelical angle than that of WT and E196K (). The 25–50 ns average WT, E196K and Y218N HB-HC interhelical angles were 131°, 133°, and 140°, respectively. In simulation 1 of Y218N, the interhelical angle increased at the beginning. The C-terminus of HB and N-terminus of HC approached each other. F198 was pushed to the semi-exposed state by HB and HC at 0.2 ns. The increase in the interhelical angle also accommodated backbone conformational changes within the HB-HC loop, which allowed F198 to become fully exposed at 18 ns. Interestingly, the native WT salt bridge between E196 and R156 was preserved in Y218N. Despite this, the capping box interaction between T199 and D202 was lost. The difference in the salt bridge network and interhelical angles between E196K and Y218N demonstrated the different pathways for disrupting of the hydrophobic packing around F198 in these two mutants.
Figure 7. Change in interhelical angle between HB and HC in Y218N. (A) Structures in simulation 1 of Y218N. Only the HB-HC loop (gray), HB and HC (blue) are shown. The red helices represent the starting structure of residues 179–194 and 200–214, where the interhelical angle is defined between those two fragments of HB and HC. Interhelical angles between HB and HC for each snapshot are indicated. (B) Inter helical angle between HB and HC at 100 ps granularity. The average interhelical angle of the last 25 ns of the WT simulations is shown in dashed lines.
Loss of X-loop hydrophobic packing in Y218N
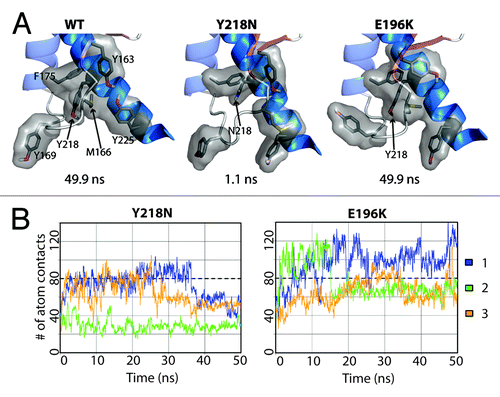
While E196K and WT displayed comparable compactness at the X-loop, Y218N lost significant X-loop packing interactions as a result of the mutation (). To measure the hydrophobic packing at the X-loop, the number of atom contacts, consisting of a list of bulky residues around the X-loop (including residue Y/N218 and M166), were quantified for all simulations (). Both WT and E196K had about 80 atom contacts and M166 was always buried between HB and HC. Y218N tended to have less atomic contacts compared with WT and E196K. Simulation 2 of Y218N had an exceptionally low number of atom contacts (~28 contacts). This was caused by the change in the X-loop conformation at 1.1 ns such that M166 became exposed to solvent.
Figure 8. Loss of hydrophobic packing around in the X-loop in Y218N simulations. (A) Relevant residues involved in X-loop packing interactions are shown in sticks for simulation 2 of WT, Y218N, and E196K (B) Number of atom contacts between residues Y163, M166, Y169, F175, Y/N218, and Y225. Window averages (100 ps) of number of atom contacts are shown for all mutant simulations. The average number of atom contacts of the last 25 ns of the WT simulations is shown in dashed lines.
Nonnative strand at the flexible N-terminus
A nonnative strand (denoted as E1) at the flexible N-terminus formed () within the first 3 ns of both Y218N and E196K simulations. Once formed, E1 was stable throughout the rest of the simulation (). E1 formed independently of other conversion events (e.g., the large HA movement and the disrupted hydrophobic core). E1 used the native strand, S1, as a nucleation site and formed ≥ 4 backbone hydrogen bonds with S1. There were stable β-bulges in E1, which formed short, α-strand-like backbone conformations that are believed to be early steps of amyloidosis.Citation33,Citation34 The residues that participated in E1 formation were in the range of 117–124, which overlap with critical amyloidogenic sequences in the N-terminus, such as the 112-AGAAAAGA-119 palindromeCitation15 and the neurotoxic peptide 106–126.Citation16,Citation35
Figure 9. Formation of nonnative strand (E1) in both Y218N and E196K simulations. (A) Representative E1 structure for each mutant. Backbone of E1 is shown in sticks with carbon atoms colored in orange. Carbon atoms of the extended native sheets are colored in red. Hydrogen bonds between strands are indicated with the cyan dashed lines. The N- and C-terminal residues for E1 and the extended S1 strand (in red) are labeled. (B) Secondary structure analysis of simulation 2 of Y218N and simulation 3 of E196K.
Discussion
Using MD simulations, we studied the effects of two pathogenic mutants Y218N and E196K on the structure and dynamics of the human PrP. The mutation sites caused local differences between the mutants. The E196K mutation eliminated the WT salt bridge E196-R156. The Y218N mutation resulted in a loss of hydrophobic packing around the X-loop region. Despite local differences around the mutation sites, the two mutants shared common traits of misfolding: 1) HA detached from PrP core; 2) F198 left its native position and became solvent exposed; and 3) the flexible N-terminus formed a new strand. Here we have shown that each mutant has a unique pathway for triggering common misfolding events.
Repositioning of the HA helix
In mutant Y218N simulations, the mutation caused HC to bend toward the S1-HA loop; E221 then formed a stable contact with S132 on the S1-HA loop (). The S132-E221 contact was only scarcely (< 1% simulation time) populated in WT simulations. This nonnative contact offered a pathway for the mutation to affect the S1-HA loop conformation. The change in the S1-HA loop conformation disrupts the hydrophobic packing between the S1-HA loop and HC, which therefore destabilizes HA. The importance of the hydrophobic packing between HC and the S1-HA loop has been verified by mutational studies on PrP: polar residue mutations of M206 and M213 reduce thermal stability and enhance amyloidogenicity of PrPC.Citation36
The native WT salt bridge E196-R156 tethers HA to the protein core. This salt bridge, which is present in NMR structures of human PrP,Citation11,Citation37 is also highly populated in several MD studies.Citation38-Citation41 Low pH MD simulations of human PrP resulted in loss of the E196-R156 salt bridge and HA detachment from HC.Citation31 The E196K mutation abolished the native WT salt bridge due to the charge repulsion between K196 and R156. However, the effect of the mutation was not observed immediately. Hydrophobic packing between the S1-HA loop and HC, and also the hydrogen bond between D202 and Y149, held HA intact until later in simulation when HA detached from HC ().
The stability of HA can be influenced by low pH,Citation42 chemical denaturants,Citation43 temperature and pressure.Citation44 Our previous MD studies first suggest that HA significantly changes its position at low pH.Citation27,Citation28,Citation31,Citation38,Citation45 Furthermore, two other groups have also used computational methods and found HA to be significantly repositioned.Citation46,Citation47 HA repositioning has also been observed in MD simulations of pathogenic PrP mutants.Citation26 These studies suggest that the repositioned HA conformation is permissible and that it can be induced under destabilizing conditions. An engineered disulfide bond (by double-cysteine mutants) that tethers HA to HC prevents formation of PrP oligomersCitation48 and fibrils in in vitro experiments.Citation49 In vivo cell studies indicate that these double-cysteine mutants do not propagate PrPSc formation.Citation49 These experimental findings suggest that separation of HA and HC is necessary for oligomerization and propagation of PrPSc, and thereby support our computational observation that repositioning of HA is an important hallmark for PrPSc conversion.
Exposure of F198 and disruption of the hydrophobic pocket between HB and HC
The E196K mutation had a direct impact on the hydrophobic core of the PrP (). F198 became fully exposed due to its steric repulsion with K196. The N-capping box interaction at HC was lost; furthermore K196 formed a stable salt bridge with D202 which prevented the side chain hydrogen bond between D202 and T199. Loss of the capping box interaction allowed for more conformational freedom of the HB-HC loop (residues 194–199), thus facilitating disruption of the F198 hydrophobic pocket.
Y218N also adapted a different HB-HC loop conformation that exposed F198 to the solvent, but by a very different mechanism. The Y218N mutation resulted in a significant loss of hydrophobic packing around the X-loop (). The mutation also caused a significant increase in the HB-HC interhelical angle (), so that HB and HC became more antiparallel to each other. As the interhelical angle increased, the hydrophobic pocket that contained F198 was disrupted and so was the HB-HC loop. The HB-HC loop adopted a nonnative conformation and F198 became exposed to solvent.
Riek et al. previously predicted that a pathogenic mutation, F198S, introduces a destabilizing void volume in the PrP core.Citation32 Subsequent thermodynamic studies are in agreement with this prediction,Citation24 and MD studies have also indicated that F198S destabilizes PrPC.Citation26,Citation50,Citation51 In vitro results confirmed that F198S has a higher tendency to misfold.Citation52,Citation53 From these studies of F198S, we conclude that the loss of hydrophobic packing around F198 facilitates misfolding. Denaturation studies indicate that stable NOE contacts exist between F198-M206 and F198-Y157 in bovine PrP,Citation53 which confirms the stabilizing effect of F198 in the native state. The binding of a PrPSc-specific antibody to residues 187–206 suggests that the loop between HB and HC adapts a misfolded/altered conformation in PrPSc.Citation54 Thus, change in the HB-HC loop conformation and exposure of F198 to solvent appears to be another likely hallmark for PrPSc conversion.
Putative aggregation sites: the flexible N-terminus and the S1-HA loop
In both mutants, we observed significant movement of HA and the S1-HA loop. The number of hydrophobic contacts between the S1-HA loop and HC were significantly reduced as HA detached from HC. Mutation studies on the PrP have confirmed that the hydrophobic packing between the S1-HA loop and HC are crucial for the native PrPC thermal stability.Citation36,Citation55 The loss of hydrophobic packing between HC and the S1-HA loop allowed higher conformational flexibility for the S1-HA. High pressure unfolding experiments performed on the PrP have also identified that residues I139, H140, and F141 in the S1-HA loop have a higher conformational variability/heterogeneity,Citation56 which might be related to the early steps of misfolding. According to studies on prion peptic fragments, the sequence of residues 136–140 is critical for aggregation.Citation57 In particular, alanine mutations at hydrophobic residues P137 and I139 completely halted conversion to PrPSc. This indicates that the hydrophobic residues in the S1-HA loop play an important role in misfolding and aggregation. Abalos et al. further hypothesized that residues 136–140 participate in the PrPSc-PrPC binding interface and also the process of misfolding.Citation57 This hypothesis is in agreement with our first simulations of PrPC conversionCitation27 and our later protofibril models.Citation28,Citation58 We hypothesize that, during PrPSc oligomerization, the S1-HA loop forms a new strand by docking with a neighboring PrPSc unit. The new strand at S1-HA loop is one of the putative aggregation site denoted as E4.Citation28
In vivo experiments show that the flexible N-terminus in PrPC is required for extracellular β-oligomers to induce cellular toxicity.Citation59,Citation60 In vitro studies indicate that residues from the flexible N-terminus are critical for aggregation, such as residues 98–110,Citation57 the 112-AGAAAAGA-119 palindrome,Citation15 neurotoxic peptide 106–126,Citation16,Citation35 polybasic region (residues 94–110),Citation18 and the mini prion 106.Citation17,Citation61 Interestingly, there is a pathogenic Y145Stop mutant,Citation62,Citation63 which suggests that a neurotoxic conformation is encoded in the PrP residues before HA. Our simulations detected the formation of the E1 strand at the flexible N-terminus, which has been observed in previous MD simulations of other pathogenic mutantsCitation26,Citation64 and also under acidic pH conditions.Citation27,Citation28,Citation31 These data suggest that the N-terminus is critical to the conversion process and must be included in simulation studies of PrP. In our spiral model for PrPSc soluble oligomers,Citation28 the PrPSc monomeric units have a nonnative strand formed at the N-terminus, which is docked to E4 (the S1-HA loop). We hypothesize that the nonnative strand formed at the N-terminus is an aggregation site for PrPSc.
Structural basis for different prion strains
Although both Y218N and E196K differ only by two residues in their primary sequences, they share significant differences in the X-loop conformation and HB-HC interhelical angle. A single point mutation (D167S) changing the structure of the X-loop in mouse PrP can cause prion disease,Citation65 but the relationship between structure, sequence and disease phenotype is still obscure. Previously, we have proposed that the differences in X-loop conformations provide a structural basis for prion disease resistance.Citation66 This is likewise applicable to explain the differences between Y218N and E196K, as they cause two different prion diseases, GSS and familial CJD, respectively. The spiral modelCitation28 places the X-loop between PrP monomer interfaces; the HB-HC helical orientation also plays a significant role in the complementarity of the PrPSc intermolecular binding surface. Antibody-binding studiesCitation67,Citation68 and X-ray diffractionCitation69,Citation70 have confirmed that distinct prion strains have different conformations. While it is premature to conclude that the differences in the X-loop conformation and HB-HC orientation would cause different pathogenesis, the significant structural differences between Y218N and E196K supports the hypothesis of a structural basis for different prion strains.
Materials and Methods
The NMR structure of human PrP,Citation11 PDB code 1QLX, was used as the starting structure for the globular domain (residues 128–228). Since residues 90–230 the critical core of PrPSc,Citation14 and the PrP fragment with residues 90–230 (PrP90–230) contains critical residues in the N-terminus that are required for PrPSc conversion,Citation15-Citation17 the missing flexible N-terminus (residues 90–127) and residues 229–230, were manually built as described previously.Citation31 Briefly, limited NOE restraints were used to model residues 90–127 such that it can move freely in any direction with respect to the globular domain. The protocol for generating starting structures of PrP mutants was described previously.Citation64 First, the side chain at the mutation site was replaced, using our rotamer libraryCitation71 built from the Dynameomics database.Citation72 Multiple structures with different rotamers at the mutation site were generated, followed by 100 steps of steepest descent minimization on all side chains. Finally, the lowest energy structure was selected as the starting structure.
Simulations were performed at neutral pH and 310 K. The protonation states of histidine residues were consistent with our previous study at neutral pH,Citation31, i.e., only H144 was protonated on Nδ1; all other histidine residues were protonated on Nε2. All side chains of aspartate and glutamate residues were negatively charged. Side chains of lysine and arginine residues were positively charged. MD simulations were performed using in lucem Molecular Mechanics (ilmm),Citation73 which uses the Levitt et al. force fieldCitation74 and F3C water model.Citation75 The NVE microcanonical ensemble was employed. Previously developed protocols were used to prepare and perform the simulations.Citation76 Triplicate simulations of 50 ns each were generated for each mutant. Four simulations of 50 ns were generated for WT. A different random number seed was used for each simulation to generate replicates to improve sampling.
Most of the analysis methods have been described previously.Citation26 The Cα RMSD of the globular domain was aligned to and calculated for residues 128–228. The Cα RMSD of HA was aligned to the stable core (residues 174–186 and 200–219) and calculated for residues 144–156. SASA was calculated by ilmm using the Lee and Richards method.Citation77 Secondary structure analysis was performed by using the DSSP (Define Secondary Structure of Proteins) algorithmCitation78 but with additional definitions.Citation79 Heavy atoms were considered to be in contact when they are less than 4.6 Å apart, or 5.4 Å apart if both heavy atoms were carbon atoms. Hydrogen bonds were defined by a 2.6 Å distance between a hydrogen atom and an acceptor and the donor-H-acceptor angle must be within 45° of linearity. In order to calculate inter-helical angle between HB and HC, Cα atoms of residues 179–194 (on HB) and 200–214 (on HC) were fit to two vectors and the angle between the two vectors was measured.
| Abbreviations: | ||
| PrPC | = | cellular form of the prion protein |
| PrPSc | = | scrapie prion protein |
| MD | = | molecular dynamics |
| CJD | = | Creutzfeldt-Jakob disease |
| GSS | = | Gerstmann-Sträussler-Scheinker Syndrome |
| GPI | = | glycosylphosphatidyl-inositol |
| WT | = | wild-type |
| Cα RMSD | = | root-mean-square-deviation of Cα atoms |
| SASA | = | solvent accessible surface area |
| DSSP | = | algorithm of defining secondary structure of proteins |
| sc | = | side chain of a residue |
| mc | = | main chain of a residue |
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We are grateful for financial support provided by the National Institutes of Health (GMS 95836 to V.D.). We thank Dr. Marc van der Kamp and Dr. Wei Chen for technical support.
Financial disclosure
This study was supported by the National Institutes of Health (GM 95836 to V.D.).
References
- Wadsworth JDF, Collinge J. Molecular pathology of human prion disease. Acta Neuropathol 2011; 121:69 - 77; http://dx.doi.org/10.1007/s00401-010-0735-5; PMID: 20694796
- Head MW, Ironside JW. Review: Creutzfeldt-Jakob disease: prion protein type, disease phenotype and agent strain. Neuropathol Appl Neurobiol 2012; 38:296 - 310; http://dx.doi.org/10.1111/j.1365-2990.2012.01265.x; PMID: 22394291
- Gambetti P, Cali I, Notari S, Kong Q, Zou W-Q, Surewicz WK. Molecular biology and pathology of prion strains in sporadic human prion diseases. Acta Neuropathol 2011; 121:79 - 90; http://dx.doi.org/10.1007/s00401-010-0761-3; PMID: 21058033
- Bett C, Joshi-Barr S, Lucero M, Trejo M, Liberski P, Kelly JW, Masliah E, Sigurdson CJ. Biochemical properties of highly neuroinvasive prion strains. PLoS Pathog 2012; 8:e1002522; http://dx.doi.org/10.1371/journal.ppat.1002522; PMID: 22319450
- Puoti G, Bizzi A, Forloni G, Safar JG, Tagliavini F, Gambetti P. Sporadic human prion diseases: molecular insights and diagnosis. Lancet Neurol 2012; 11:618 - 28; http://dx.doi.org/10.1016/S1474-4422(12)70063-7; PMID: 22710755
- Borchelt DR, Scott M, Taraboulos A, Stahl N, Prusiner SB. Scrapie and cellular prion proteins differ in their kinetics of synthesis and topology in cultured cells. J Cell Biol 1990; 110:743 - 52; http://dx.doi.org/10.1083/jcb.110.3.743; PMID: 1968466
- Caughey B, Raymond GJ. The scrapie-associated form of PrP is made from a cell surface precursor that is both protease- and phospholipase-sensitive. J Biol Chem 1991; 266:18217 - 23; PMID: 1680859
- James TL, Liu H, Ulyanov NB, Farr-Jones S, Zhang H, Donne DG, Kaneko K, Groth D, Mehlhorn I, Prusiner SB, et al. Solution structure of a 142-residue recombinant prion protein corresponding to the infectious fragment of the scrapie isoform. Proc Natl Acad Sci U S A 1997; 94:10086 - 91; http://dx.doi.org/10.1073/pnas.94.19.10086; PMID: 9294167
- Riek R, Hornemann S, Wider G, Billeter M, Glockshuber R, Wüthrich K. NMR structure of the mouse prion protein domain PrP(121-231). Nature 1996; 382:180 - 2; http://dx.doi.org/10.1038/382180a0; PMID: 8700211
- López Garcia F, Zahn R, Riek R, Wüthrich K. NMR structure of the bovine prion protein. Proc Natl Acad Sci U S A 2000; 97:8334 - 9; http://dx.doi.org/10.1073/pnas.97.15.8334; PMID: 10899999
- Zahn R, Liu A, Lührs T, Riek R, von Schroetter C, López García F, Billeter M, Calzolai L, Wider G, Wüthrich K. NMR solution structure of the human prion protein. Proc Natl Acad Sci U S A 2000; 97:145 - 50; http://dx.doi.org/10.1073/pnas.97.1.145; PMID: 10618385
- Stöhr J, Watts JC, Legname G, Oehler A, Lemus A, Nguyen H-OB, Sussman J, Wille H, DeArmond SJ, Prusiner SB, et al. Spontaneous generation of anchorless prions in transgenic mice. Proc Natl Acad Sci U S A 2011; 108:21223 - 8; http://dx.doi.org/10.1073/pnas.1117827108; PMID: 22160704
- Chesebro B, Trifilo M, Race R, Meade-White K, Teng C, LaCasse R, Raymond L, Favara C, Baron G, Priola S, et al. Anchorless prion protein results in infectious amyloid disease without clinical scrapie. Science 2005; 308:1435 - 9; http://dx.doi.org/10.1126/science.1110837; PMID: 15933194
- Zou W-Q, Capellari S, Parchi P, Sy M-S, Gambetti P, Chen SG. Identification of novel proteinase K-resistant C-terminal fragments of PrP in Creutzfeldt-Jakob disease. J Biol Chem 2003; 278:40429 - 36; http://dx.doi.org/10.1074/jbc.M308550200; PMID: 12917418
- Norstrom EM, Mastrianni JA. The AGAAAAGA palindrome in PrP is required to generate a productive PrPSc-PrPC complex that leads to prion propagation. J Biol Chem 2005; 280:27236 - 43; http://dx.doi.org/10.1074/jbc.M413441200; PMID: 15917252
- Jobling MF, Stewart LR, White AR, McLean C, Friedhuber A, Maher F, Beyreuther K, Masters CL, Barrow CJ, Collins SJ, et al. The hydrophobic core sequence modulates the neurotoxic and secondary structure properties of the prion peptide 106-126. J Neurochem 1999; 73:1557 - 65; http://dx.doi.org/10.1046/j.1471-4159.1999.0731557.x; PMID: 10501201
- Supattapone S, Bosque P, Muramoto T, Wille H, Aagaard C, Peretz D, Nguyen H-OB, Heinrich C, Torchia M, Safar J, et al. Prion protein of 106 residues creates an artifical transmission barrier for prion replication in transgenic mice. Cell 1999; 96:869 - 78; http://dx.doi.org/10.1016/S0092-8674(00)80596-6; PMID: 10102274
- Solomon IH, Khatri N, Biasini E, Massignan T, Huettner JE, Harris DA. An N-terminal polybasic domain and cell surface localization are required for mutant prion protein toxicity. J Biol Chem 2011; 286:14724 - 36; http://dx.doi.org/10.1074/jbc.M110.214973; PMID: 21385869
- Legname G, Baskakov IV, Nguyen H-OB, Riesner D, Cohen FE, DeArmond SJ, Prusiner SB. Synthetic mammalian prions. Science 2004; 305:673 - 6; http://dx.doi.org/10.1126/science.1100195; PMID: 15286374
- van der Kamp MW, Daggett V. The consequences of pathogenic mutations to the human prion protein. Protein Eng Des Sel 2009; 22:461 - 8; http://dx.doi.org/10.1093/protein/gzp039; PMID: 19602567
- Lloyd SE, Mead S, Collinge J. Genetics of prion diseases. Curr Opin Genet Dev 2013; 23:345 - 51; http://dx.doi.org/10.1016/j.gde.2013.02.012; PMID: 23518043
- Beck JA, Poulter M, Campbell TA, Adamson G, Uphill JB, Guerreiro R, Jackson GS, Stevens JC, Manji H, Collinge J, et al. PRNP allelic series from 19 years of prion protein gene sequencing at the MRC Prion Unit. Hum Mutat 2010; 31:E1551 - 63; http://dx.doi.org/10.1002/humu.21281; PMID: 20583301
- Giachin G, Biljan I, Ilc G, Plavec J, Legname G. Probing early misfolding events in prion protein mutants by NMR spectroscopy. Molecules 2013; 18:9451 - 76; http://dx.doi.org/10.3390/molecules18089451; PMID: 23966072
- Liemann S, Glockshuber R. Influence of amino acid substitutions related to inherited human prion diseases on the thermodynamic stability of the cellular prion protein. Biochemistry 1999; 38:3258 - 67; http://dx.doi.org/10.1021/bi982714g; PMID: 10079068
- Swietnicki W, Petersen RB, Gambetti P, Surewicz WK. Familial mutations and the thermodynamic stability of the recombinant human prion protein. J Biol Chem 1998; 273:31048 - 52; http://dx.doi.org/10.1074/jbc.273.47.31048; PMID: 9813003
- van der Kamp MW, Daggett V. Pathogenic mutations in the hydrophobic core of the human prion protein can promote structural instability and misfolding. J Mol Biol 2010; 404:732 - 48; http://dx.doi.org/10.1016/j.jmb.2010.09.060; PMID: 20932979
- Alonso DOV, DeArmond SJ, Cohen FE, Daggett V. Mapping the early steps in the pH-induced conformational conversion of the prion protein. Proc Natl Acad Sci U S A 2001; 98:2985 - 9; http://dx.doi.org/10.1073/pnas.061555898; PMID: 11248018
- DeMarco ML, Daggett V. From conversion to aggregation: protofibril formation of the prion protein. Proc Natl Acad Sci U S A 2004; 101:2293 - 8; http://dx.doi.org/10.1073/pnas.0307178101; PMID: 14983003
- Alzualde A, Indakoetxea B, Ferrer I, Moreno F, Barandiaran M, Gorostidi A, Estanga A, Ruiz I, Calero M, van Leeuwen FW, et al. A novel PRNP Y218N mutation in Gerstmann-Sträussler-Scheinker disease with neurofibrillary degeneration. J Neuropathol Exp Neurol 2010; 69:789 - 800; http://dx.doi.org/10.1097/NEN.0b013e3181e85737; PMID: 20613639
- Peoc’h K, Manivet P, Beaudry P, Attane F, Besson G, Hannequin D, Delasnerie-Lauprêtre N, Laplanche J-L. Identification of three novel mutations (E196K, V203I, E211Q) in the prion protein gene (PRNP) in inherited prion diseases with Creutzfeldt-Jakob disease phenotype. Hum Mutat 2000; 15:482; http://dx.doi.org/10.1002/(SICI)1098-1004(200005)15:5<482::AID-HUMU16>3.0.CO;2-1; PMID: 10790216
- van der Kamp MW, Daggett V. Influence of pH on the human prion protein: insights into the early steps of misfolding. Biophys J 2010; 99:2289 - 98; http://dx.doi.org/10.1016/j.bpj.2010.07.063; PMID: 20923664
- Riek R, Wider G, Billeter M, Hornemann S, Glockshuber R, Wüthrich K. Prion protein NMR structure and familial human spongiform encephalopathies. Proc Natl Acad Sci U S A 1998; 95:11667 - 72; http://dx.doi.org/10.1073/pnas.95.20.11667; PMID: 9751723
- Daggett V. Alpha-sheet: The toxic conformer in amyloid diseases?. Acc Chem Res 2006; 39:594 - 602; http://dx.doi.org/10.1021/ar0500719; PMID: 16981675
- Armen RS, DeMarco ML, Alonso DOV, Daggett V. Pauling and Corey’s alpha-pleated sheet structure may define the prefibrillar amyloidogenic intermediate in amyloid disease. Proc Natl Acad Sci U S A 2004; 101:11622 - 7; http://dx.doi.org/10.1073/pnas.0401781101; PMID: 15280548
- Forloni G, Angeretti N, Chiesa R, Monzani E, Salmona M, Bugiani O, Tagliavini F. Neurotoxicity of a prion protein fragment. Nature 1993; 362:543 - 6; http://dx.doi.org/10.1038/362543a0; PMID: 8464494
- Lisa S, Meli M, Cabello G, Gabizon R, Colombo G, Gasset M. The structural intolerance of the PrP alpha-fold for polar substitution of the helix-3 methionines. Cell Mol Life Sci 2010; 67:2825 - 38; http://dx.doi.org/10.1007/s00018-010-0363-1; PMID: 20454997
- Calzolai L, Zahn R. Influence of pH on NMR structure and stability of the human prion protein globular domain. J Biol Chem 2003; 278:35592 - 6; http://dx.doi.org/10.1074/jbc.M303005200; PMID: 12826672
- DeMarco ML, Daggett V. Molecular mechanism for low pH triggered misfolding of the human prion protein. Biochemistry 2007; 46:3045 - 54; http://dx.doi.org/10.1021/bi0619066; PMID: 17315950
- Zuegg J, Gready JE. Molecular dynamics simulations of human prion protein: importance of correct treatment of electrostatic interactions. Biochemistry 1999; 38:13862 - 76; http://dx.doi.org/10.1021/bi991469d; PMID: 10529232
- Bamdad K, Naderi-Manesh H. Contribution of a putative salt bridge and backbone dynamics in the structural instability of human prion protein upon R208H mutation. Biochem Biophys Res Commun 2007; 364:719 - 24; http://dx.doi.org/10.1016/j.bbrc.2007.10.011; PMID: 17980350
- Langella E, Improta R, Crescenzi O, Barone V. Assessing the acid-base and conformational properties of histidine residues in human prion protein (125-228) by means of pK(a) calculations and molecular dynamics simulations. Proteins 2006; 64:167 - 77; http://dx.doi.org/10.1002/prot.20979; PMID: 16639746
- Watanabe Y, Inanami O, Horiuchi M, Hiraoka W, Shimoyama Y, Inagaki F, Kuwabara M. Identification of pH-sensitive regions in the mouse prion by the cysteine-scanning spin-labeling ESR technique. Biochem Biophys Res Commun 2006; 350:549 - 56; http://dx.doi.org/10.1016/j.bbrc.2006.09.082; PMID: 17022940
- Hosszu LLP, Wells MA, Jackson GS, Jones S, Batchelor M, Clarke AR, Craven CJ, Waltho JP, Collinge J. Definable equilibrium states in the folding of human prion protein. Biochemistry 2005; 44:16649 - 57; http://dx.doi.org/10.1021/bi051277k; PMID: 16342955
- Torrent J, Alvarez-Martinez MT, Liautard J-P, Balny C, Lange R. The role of the 132-160 region in prion protein conformational transitions. Protein Sci 2005; 14:956 - 67; http://dx.doi.org/10.1110/ps.04989405; PMID: 15772306
- Alonso DOV, An C, Daggett V. Simulations of biomolecules: Characterization of the early steps in the pH-induced conformational conversion of the hamster, bovine and human forms of the prion protein. Eng Sci 2002; 360:1165 - 78
- Colacino S, Tiana G, Broglia RA, Colombo G. The determinants of stability in the human prion protein: insights into folding and misfolding from the analysis of the change in the stabilization energy distribution in different conditions. Proteins 2006; 62:698 - 707; http://dx.doi.org/10.1002/prot.20804; PMID: 16432880
- De Simone A, Zagari A, Derreumaux P. Structural and hydration properties of the partially unfolded states of the prion protein. Biophys J 2007; 93:1284 - 92; http://dx.doi.org/10.1529/biophysj.107.108613; PMID: 17483173
- Eghiaian F, Daubenfeld T, Quenet Y, van Audenhaege M, Bouin A-P, van der Rest G, Grosclaude J, Rezaei H. Diversity in prion protein oligomerization pathways results from domain expansion as revealed by hydrogen/deuterium exchange and disulfide linkage. Proc Natl Acad Sci U S A 2007; 104:7414 - 9; http://dx.doi.org/10.1073/pnas.0607745104; PMID: 17442756
- Hafner-Bratkovic I, Bester R, Pristovšek P, Gaedtke L, Veranič P, Gašperšič J, Manček-Keber M, Avbelj M, Polymenidou M, Julius C, et al. Globular domain of the prion protein needs to be unlocked by domain swapping to support prion protein conversion. J Biol Chem 2011; 286:12149 - 56; http://dx.doi.org/10.1074/jbc.M110.213926; PMID: 21324909
- Zhang Y, Qian J, Wang P-Y, Ou-Yang Z-C. Molecular dynamical simulations of point mutation occurring at the 198-th site of prion protein. J Comput Theor Nanosci 2006; 3:964 - 9; http://dx.doi.org/10.1166/jctn.2006.012
- Zhang Y, Dai L, Iwamoto M, Ou-Yang Z. Molecular dynamics study on the conformational transition of prion induced by the point mutation: F198S. Thin Solid Films 2006; 499:224 - 8; http://dx.doi.org/10.1016/j.tsf.2005.07.008
- Vanik DL, Surewicz WK. Disease-associated F198S mutation increases the propensity of the recombinant prion protein for conformational conversion to scrapie-like form. J Biol Chem 2002; 277:49065 - 70; http://dx.doi.org/10.1074/jbc.M207511200; PMID: 12372829
- Julien O, Chatterjee S, Thiessen A, Graether SP, Sykes BD. Differential stability of the bovine prion protein upon urea unfolding. Protein Sci 2009; 18:2172 - 82; http://dx.doi.org/10.1002/pro.231; PMID: 19693935
- Jones M, McLoughlin V, Connolly JG, Farquhar CF, MacGregor IR, Head MW. Production and characterization of a panel of monoclonal antibodies against native human cellular prion protein. Hybridoma (Larchmt) 2009; 28:13 - 20; http://dx.doi.org/10.1089/hyb.2008.0067; PMID: 19132894
- Hart T, Hosszu LLP, Trevitt CR, Jackson GS, Waltho JP, Collinge J, Clarke AR. Folding kinetics of the human prion protein probed by temperature jump. Proc Natl Acad Sci U S A 2009; 106:5651 - 6; http://dx.doi.org/10.1073/pnas.0811457106; PMID: 19321423
- Kachel N, Kremer W, Zahn R, Kalbitzer HR. Observation of intermediate states of the human prion protein by high pressure NMR spectroscopy. BMC Struct Biol 2006; 6:16; http://dx.doi.org/10.1186/1472-6807-6-16; PMID: 16846506
- Abalos GC, Cruite JT, Bellon A, Hemmers S, Akagi J, Mastrianni JA, Williamson RA, Solforosi L. Identifying key components of the PrPC-PrPSc replicative interface. J Biol Chem 2008; 283:34021 - 8; http://dx.doi.org/10.1074/jbc.M804475200; PMID: 18826953
- DeMarco ML, Silveira J, Caughey B, Daggett V. Structural properties of prion protein protofibrils and fibrils: an experimental assessment of atomic models. Biochemistry 2006; 45:15573 - 82; http://dx.doi.org/10.1021/bi0612723; PMID: 17176078
- Resenberger UK, Harmeier A, Woerner AC, Goodman JL, Müller V, Krishnan R, Vabulas RM, Kretzschmar HA, Lindquist S, Hartl FU, et al. The cellular prion protein mediates neurotoxic signalling of β-sheet-rich conformers independent of prion replication. EMBO J 2011; 30:2057 - 70; http://dx.doi.org/10.1038/emboj.2011.86; PMID: 21441896
- Rambold AS, Müller V, Ron U, Ben-Tal N, Winklhofer KF, Tatzelt J. Stress-protective signalling of prion protein is corrupted by scrapie prions. EMBO J 2008; 27:1974 - 84; http://dx.doi.org/10.1038/emboj.2008.122; PMID: 18566584
- Supattapone S, Bouzamondo E, Ball HL, Wille H, Nguyen H-OB, Cohen FE, DeArmond SJ, Prusiner SB, Scott M. A protease-resistant 61-residue prion peptide causes neurodegeneration in transgenic mice. Mol Cell Biol 2001; 21:2608 - 16; http://dx.doi.org/10.1128/MCB.21.7.2608-2616.2001; PMID: 11259607
- Kundu B, Maiti NR, Jones EM, Surewicz KA, Vanik DL, Surewicz WK. Nucleation-dependent conformational conversion of the Y145Stop variant of human prion protein: structural clues for prion propagation. Proc Natl Acad Sci U S A 2003; 100:12069 - 74; http://dx.doi.org/10.1073/pnas.2033281100; PMID: 14519851
- Helmus JJ, Surewicz K, Nadaud PS, Surewicz WK, Jaroniec CP. Molecular conformation and dynamics of the Y145Stop variant of human prion protein in amyloid fibrils. Proc Natl Acad Sci U S A 2008; 105:6284 - 9; http://dx.doi.org/10.1073/pnas.0711716105; PMID: 18436646
- Chen W, van der Kamp MW, Daggett V. Diverse effects on the native β-sheet of the human prion protein due to disease-associated mutations. Biochemistry 2010; 49:9874 - 81; http://dx.doi.org/10.1021/bi101449f; PMID: 20949975
- Sigurdson CJ, Joshi-Barr S, Bett C, Winson O, Manco G, Schwarz P, Rülicke T, Nilsson KPR, Margalith I, Raeber A, et al. Spongiform encephalopathy in transgenic mice expressing a point mutation in the β2-α2 loop of the prion protein. J Neurosci 2011; 31:13840 - 7; http://dx.doi.org/10.1523/JNEUROSCI.3504-11.2011; PMID: 21957246
- Scouras AD, Daggett V. Disruption of the X-loop turn of the prion protein linked to scrapie resistance. Protein Eng Des Sel 2012; 25:243 - 9; http://dx.doi.org/10.1093/protein/gzs009; PMID: 22447804
- Safar J, Wille H, Itri V, Groth D, Serban H, Torchia M, Cohen FE, Prusiner SB. Eight prion strains have PrP(Sc) molecules with different conformations. Nat Med 1998; 4:1157 - 65; http://dx.doi.org/10.1038/2654; PMID: 9771749
- Nilsson KPR, Joshi-Barr S, Winson O, Sigurdson CJ. Prion strain interactions are highly selective. J Neurosci 2010; 30:12094 - 102; http://dx.doi.org/10.1523/JNEUROSCI.2417-10.2010; PMID: 20826672
- Wille H, Bian W, McDonald M, Kendall A, Colby DW, Bloch L, Ollesch J, Borovinskiy AL, Cohen FE, Prusiner SB, et al. Natural and synthetic prion structure from X-ray fiber diffraction. Proc Natl Acad Sci U S A 2009; 106:16990 - 5; http://dx.doi.org/10.1073/pnas.0909006106; PMID: 19805070
- Makarava N, Kovacs GG, Savtchenko R, Alexeeva I, Budka H, Rohwer RG, Baskakov IV. Genesis of mammalian prions: from non-infectious amyloid fibrils to a transmissible prion disease. PLoS Pathog 2011; 7:e1002419; http://dx.doi.org/10.1371/journal.ppat.1002419; PMID: 22144901
- Scouras AD, Daggett V. The Dynameomics rotamer library: amino acid side chain conformations and dynamics from comprehensive molecular dynamics simulations in water. Protein Sci 2011; 20:341 - 52; http://dx.doi.org/10.1002/pro.565; PMID: 21280126
- van der Kamp MW, Schaeffer RD, Jonsson AL, Scouras AD, Simms AM, Toofanny RD, Benson NC, Anderson PC, Merkley ED, Rysavy S, et al. Dynameomics: a comprehensive database of protein dynamics. Structure 2010; 18:423 - 35; http://dx.doi.org/10.1016/j.str.2010.01.012; PMID: 20399180
- Beck DAC, McCully ME, Alonso DOV, Daggett V. in lucem Molecular Mechanics (ilmm). University of Washington, Seattle: 2000-2014.
- Levitt M, Hirshberg M, Sharon R, Daggett V. Potential energy function and parameters for simulations of the molecular dynamics of proteins and nucleic acids in solution. Comput Phys Commun 1995; 91:215 - 31; http://dx.doi.org/10.1016/0010-4655(95)00049-L
- Levitt M, Hirshberg M, Sharon R, Laidig KE, Daggett V. Calibration and testing of a water model for simulation of the molecular dynamics of proteins and nucleic acids in solution. J Phys Chem B 1997; 101:5051 - 61; http://dx.doi.org/10.1021/jp964020s
- Beck DAC, Daggett V. Methods for molecular dynamics simulations of protein folding/unfolding in solution. Methods 2004; 34:112 - 20; http://dx.doi.org/10.1016/j.ymeth.2004.03.008; PMID: 15283920
- Lee B, Richards FM. The interpretation of protein structures: estimation of static accessibility. J Mol Biol 1971; 55:379 - 400; http://dx.doi.org/10.1016/0022-2836(71)90324-X; PMID: 5551392
- Kabsch W, Sander C. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 1983; 22:2577 - 637; http://dx.doi.org/10.1002/bip.360221211; PMID: 6667333
- Scouras AD, Daggett V. Species variation in PrPSc protofibril models. J Mater Sci 2008; 43:3625 - 37; http://dx.doi.org/10.1007/s10853-008-2578-1