
Abstract
The cellular prion protein (PrPC) is an N-glycosylated GPI-anchored protein usually present in lipid rafts with numerous putative functions. When it changes its conformation to a pathological isoform (then referred to as PrPSc), it is an essential part of the prion, the agent causing fatal and transmissible neurodegenerative prion diseases. There is growing evidence that toxicity and neuronal damage on the one hand and propagation/infectivity on the other hand are two distinct processes of the disease and that the GPI-anchor attachment of PrPC and PrPSc plays an important role in protein localization and in neurotoxicity. Here we review how the signal sequence of the GPI-anchor matters in PrPC localization, how an altered cellular localization of PrPC or differences in GPI-anchor composition can affect prion infection, and we discuss through which mechanisms changes on the anchorage of PrPC can modify the disease process.
PrP, a GPI-Anchored Protein
The prion protein (PrPC) acquires two post-translational modifications along the secretory pathway: the addition of up to two N-glycan moieties at positions 181 and 197 (human amino acid numbering) and a glycosylphosphatidylinositol (GPI)-anchor at position 230 which attaches the protein to the outer leaflet of the plasma membrane (for review see ref. Citation1). Through a yet not fully understood mechanism, PrPC, which is highly expressed in neurons, can change its conformation into an abnormal protease-resistant, potentially infectious isoform (PrPSc). The latter is an essential part of the agent responsible for transmissible spongiform encephalopathies (TSE), the prion, and is still GPI-anchored.Citation2,Citation3 In this review, to simplify matters, we will use the term PrPSc as a general term for disease-associated PrP species independently of their biochemical properties or aggregation status, although it should be noted that different terms exist that imply conceptual differences.
The GPI-anchorage is highly conserved among eukaryotes and used by 10–20% of all membrane proteins in eukaryotic cells. Biosynthetically it is a complex attachment involving more than 20 genes implicated in ten reactions in the endoplasmatic reticulum (ER) which are not yet fully understood.Citation4,Citation5 GPI-anchored proteins (GPI-APs) have a wide range of functions ranging from receptors and cell surface hydrolases to adhesion molecules among othersCitation6,Citation7 but there is no clear correlation between GPI-APs and distinct biological functions. Therefore it is hypothesized that the GPI-anchor bestows some advantages over other types of more simple attachments. For instance, GPI-APs are more mobile within the plane of the lipid bilayer and occupy less space than proteins with a transmembrane domain, allowing the proteins to be more tightly packed together. Moreover, GPI-APs also have the capacity of modifying the lipid composition of the membrane itselfCitation8 and GPI-anchors may also be necessary to complete the formation of the functional structure of the protein as described for Thy-1, another GPI-AP highly expressed in neurons.Citation9,Citation10 One intriguing capacity of GPI-APs is their ability to be spontaneously inserted into membranes in vitro and in vivo, a phenomenon known as “cell surface painting,”Citation11,Citation12 which has also been observed for PrPC.Citation13
All GPI-APs contain a signal sequence at the N-terminus that directs the protein to the ER during synthesis at the ribosome. Once it is translocated to the ER, a second hydrophobic signal sequence (SS-GPI) present at the C-terminus of the protein is removed by a transamidase in a fast reaction of about a minute and a pre-formed GPI-anchor is attached.Citation14 All GPI-anchors contain a common structure composed of an ethanolamine phosphate in an amide linkage to the C-terminal carboxyl group of the protein, a highly conserved glycan core of three mannose residues, glucosamine and a phosphatidylinositol (PI) group, and phospholipid tails that attach the GPI-anchor to the cell membrane. The addition of side-branching sugars and modifications of the lipid moiety during the passage through the secretory pathway to the plasma membrane depends on the type of cell and organism, thus leading to a large structural diversity. Of note, PrPC is one of only three proteins (the others being porcine membrane dipeptidaseCitation15 and CD59Citation16) known to date that contain a sialic acid in their GPI-anchor structuresCitation17 (for a review see ref. Citation18).
The Signal Sequence of GPI (SS-GPI) as a Sorting Signal
Interestingly, further lipid remodeling of the GPI-anchor occurs in the Golgi apparatus where the GPI-APs associate to specialized microdomains known as lipid rafts or detergent resistant membranes (DRMs). These domains are described as nanodomains highly enriched in sphingolipids and sterols that contain specific proteins and that can coalesce by lipid-lipid, protein-lipid, and protein-protein interactionsCitation19 forming a platform for signal transduction through cellular receptors.Citation20-Citation23 In fact, several GPI-APs, which are attached to the outer leaflet of the plasma membrane, can signal through tyrosine kinases after raft clustering,Citation24 probably mediated by transmembrane proteins that are also found in lipid rafts.Citation25
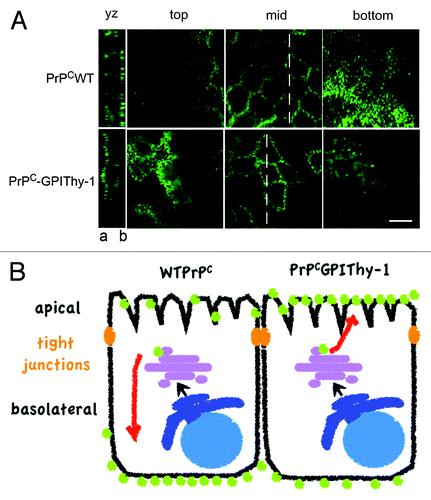
It has been proposed that the GPI-anchor is a sorting signal for selective targeting to lipid rafts and, in the majority of instances, a sorting signal for being apically sorted in polarized cells and to axons in neurons.Citation26-Citation29 However, some exceptions to this rule have been described and, in fact, PrPC is one of these exceptions, being sorted mainly basolaterally in Madin-Darby canine kidney cells (MDCK), which represent a well-established model of polarized cells.Citation30 Concerning signals that direct PrPC to the basolateral membrane, Paladino et al. proposed that the SS-GPI could play a role in its sorting. Thus, when the green fluorescent protein (GFP) is attached to the SS-GPI of PrPC, it is sorted basolaterally whereas when it is attached to the SS-GPI of the folate receptor, it is directed to the apical side in MDCK cells. The same authors also suggested that the differential mechanism for apical vs. basolateral sorting of GPI-APs in lipid rafts could be the ability of the apically sorted GPI-APs to form high molecular mass complexes (HMM) in the Golgi apparatus,Citation31 which could depend on the SS-GPI.Citation32 As shown in , our studies also revealed that, when the SS-GPI of PrPC is replaced by the one of Thy-1, the resulting PrPCGPIThy-1 relocates from the basolateral to the apical side in MDCK cells.Citation33PrPC and Thy-1 both reside in lipid rafts but share different lipid environments within these domains speaking in favor of structurally heterogeneous raft domainsCitation34,Citation35 which could have a role in sorting through HMM formation.Citation36 The localization of PrPC in neurons is more controversial. Here it can be found enriched in either axons or cell bodies and dendrites.Citation35,Citation37,Citation38 How a signal peptide, that is removed in less than a minute after the protein is translocated into the ER, can affect the sorting of GPI-APs and maybe the GPI-anchor structure and/or the lipid environment, is a question that remains unanswered. However, other factors apart from membrane composition can play a role. It is known that the SS-GPI markedly influences the efficiency of the transamidation process, which in turn may influence membrane localization.Citation11,Citation39 In a more daring concept it has also been proposed that different signal sequences can actually direct the addition of different types of GPI-anchors.Citation40,Citation41 However, experimental proof of such a mechanism is still required.
Figure 1. PrPC and PrPCGPIThy-1 expression in MDCK cells. (A) Confocal microscopy showing Z-stacks taken from top to bottom. As published beforeCitation33PrPC (in green) is basolaterally (b) sorted in fully polarized MDCK cells whereas changing the SS-GPI of PrPC for the one of Thy-1 preferentially directs the PrPCGPIThy-1 to the apical (a) compartment. Scale bar is 10μm. Originally published in PLoS One. (B) Schematic representation of the differential sorting of wild type (WT) PrPC and PrPCGPIThy-1 shown in A.
It is important to note that three point mutations (M232R, M232T, and P238S) located at the SS-GPI of PrPC lead to familial prion disease in humans.Citation42 It has been proposed that the two first mutations are related to an increased transmembrane orientation which could explain neurotoxicity.Citation43 More recently, Guizzunti et al. used a chimeric protein expressing EGFP with the SS-GPI of PrP linked to a Myc tag to demonstrate that the P238S mutation in the SS-GPI of PrP prevents degradation of the SS-GPI by the proteasome. As a consequence the cleaved SS-GPI peptide tends to accumulate in the ER. With this elegant approach, the authors monitored for the first time the fate of a GPI-anchor signal peptide and also provided an hypothesis of how mutations in the SS-GPI of PrPC might contribute to the development of prion disease.Citation44
The GPI-Anchor of PrPC and its Role in Prion Disease
The type of membrane attachment or modifications in the GPI-anchor of PrPC has an impact in the pathophysiology of prion disease. The site of conversion of PrPC to PrPSc has been proposed to be at the plasma membrane and/or in the endocytic compartmentCitation45-Citation47 with the requirement that PrPC and PrPSc are attached to the same raft domain at the membrane in cell-free conversion reactions.Citation48 A recent study using tagged PrPC, which is able to be converted to PrPSc, pinpointed de novo formation of PrPSc at the plasma membrane to lipid raft-enriched domains. Conspicuously, this is a very fast process taking about a minute. PrPSc is then rapidly internalized into early endosomes.Citation49,Citation50 These studies imply that any perturbation of lipid rafts will have an effect on prion conversion. For instance, depletion of cholesterol, either by inhibiting its biosynthesisCitation51 or by addition of amphotericin B or filipin treatment (antibiotics known to sequester cholesterol), has been shown to decrease or inhibit PrPSc formation.Citation52,Citation53 On the other hand, when PrPC is recombinantly expressed as a transmembrane protein (CD4PrPC), which is then targeted out of lipid rafts, PrPSc formation is also abolished.Citation54 Moreover, although GPI-anchorless PrPC can be efficiently converted to PrPSc in a cell-free conversion assay,Citation55 cells expressing GPI-anchorless PrPC do not support persistent infection.Citation56 Thus, in vitro, the presence of PrPC at the cell surface is a prerequisite for efficient conversion to PrPSc.Citation57
Taken together, these reports indicate that PrPC localization in lipid raft-enriched domains is of high importance for prion conversion. Interestingly, only when the amyloidogenic yeast protein Sup35 is attached to a GPI-anchor, it can be propagated to neighboring cells and these cells can then sustain and propagate the aggregates for many passages. This may raise the idea that prion diseases, in contrast to other protein misfolding diseases such as Alzheimer disease, are naturally transmissible because PrPC is the only amyloidogenic protein carrying a GPI-anchor.Citation58
Recent reports highlighted the idea that prion infection can be modified not only by the presence or absence of its GPI-anchor or by disrupting lipid rafts but also by altering the structure/composition of the GPI-anchor itself. Nisbet et al. showed that by exchanging amino acids at the C-terminal moiety within the SS-GPI of murine PrPC (MoPrP) by the ones of rabbit PrPC (RbPrP, rabbits are naturally relatively resistant to prion infectionCitation59), the resulting MoPrP-RbGPI becomes resistant to PrPSc conversion.Citation60
Interestingly, studies coming from the group of Bate and Williams showed that modifications in the lipid part of the GPI-anchor or of the sialic acid residue lead to alterations in cholesterol at the plasma membrane and to reduced PrPSc loads and infectivity (these experiments are discussed below in the section “Signaling through PrPC”).
In vivo, the scenario becomes more complex. Neuropathologically, prion diseases are characterized by the presence of diffuse or (more rarely) amyloidic depositions of PrPSc, neuropil vacuolation (spongiosis) as well as abundant astrogliosis and microglial activation. On the ultrastructural level, many of the lesions that are specific for TSE and co-localize with PrPSc are related to lesions of the plasma membrane.Citation61
Although the presence of PrPC is mandatory in order to develop prion diseases,Citation62 in inducible knockout mice infected with prions, the pathological changes in the brain are reverted once PrPC production is shut off, in spite of further PrPSc accumulation.Citation63 This is very interesting because there is growing evidence that in prion diseases there are two mechanistically different, yet related phenomena, the conversion of PrPC to PrPSc (prion propagation) and the neurotoxic signaling.Citation64 The latter is probably dependent on membrane attachment of PrPC via its GPI-anchor and on the localization of PrPC within lipid rafts as discussed later.
To study the issue of GPI-anchor attachment in vivo, Chesebro and colleagues generated mice expressing GPI-anchorless PrPC and infected them with mouse-adapted prions. They showed that GPI-anchorless PrPC is neither present in lipid rafts nor at the plasma membrane but that it is able to be converted to PrPSc, which presents with abundant thioflavin S-positive PrPSc amyloid plaques, especially around blood vessels. In these mice, infectivity was preserved but clinical onset of disease was delayed. When the animals died of disease, neurodegeneration was attributed to direct damage by amyloidic PrPSc accumulation either by disrupting the brain tissue or by causing vascular damage.Citation65Interestingly, the mice displayed a conspicuous lack of spongiosis. Spongiotic lesions can occur in a variety of neurodegenerative conditions but the type of vacuoles seen in TSE is very specific and genuine. The vacuole membrane is fragmented and contains membranes and granular osmiophilic granules.Citation61These changes are independent of PrPSc deposition. The lack of spongiotic changes in the GPI-anchorless mice was attributed either to a decreased toxicity of amyloidic anchorless PrPSc or to the absence of the GPI-anchor and thus to the lack of neurotoxic events. Our own results also point in this direction as we have generated transgenic animals expressing PrPC with the SS-GPI of Thy1 (PrPCGPIThy-1) and we have observed that there is a relocalization of PrPCGPIThy-1 to the axons. Moreover, after prion-infection, there is a delayed onset to clinical disease accompanied by an altered neuropathology, different prion strain characteristics, and decreased amounts of PrPSc (Puig et al., unpublished results).
Taken all of this into consideration, an important question arises: what are the mechanisms through which the alteration of membrane composition or alterations in the GPI-anchorage lead to changes in prion conversion and neurotoxicity? Regarding prion conversion, for GPI-anchorless PrPC it is clear that conversion is enhanced (as has also been shown in cell culture modelsCitation48) but neurotoxicity is decreased. In other instances, such as in our PrPCGPIThy-1 transgenic mice, when PrPC is localized in another cellular compartment, it is possible that direct interaction between PrPC and PrPSc is reduced at the plasma membrane and that conversion is thus slowed down. For the conversion of PrPC to PrPSc a cofactor is considered necessary (for review see ref. Citation66). Many proteins have been proposed to interact with PrPC but in vitro-data suggest that glypican-1, an heparan sulfate proteoglycan (HSPG) resident in lipid rafts, actually acts as a scaffolding protein that keeps PrPC and PrPSc in lipid rafts and facilitates prion conversion.Citation67 Therefore, meeting specific partners/cofactors in the appropriate lipid raft domain may be of importance for efficient PrPSc formation. One can also hypothesize that the endocytic pathway, necessary for the prion replication, is also altered. In fact, the mechanism of internalization of PrPC is different compared with other GPI-anchored proteins, which are usually endocytosed via a clathrin-independent pathway, although this largely depends on the cell type.Citation68PrPC leaves raft domains in order to be endocytosed via clathrin-coated pits and is rapidly endocytosed in primary neurons. Thy-1, in contrast, is internalized with a much slower rate and via non-coated vesicles.Citation69 The low-density lipoprotein receptor-related protein 1 (LRP1) is thought to be the transmembrane protein that supports PrPC to be internalized via clathrin-coated pits.Citation70,Citation71 Other mechanisms implicating cooperation between lipid rafts and clathrin have also been proposed.Citation72 One might speculate that by changing either the properties of the GPI-anchor or the membrane composition, endocytosis might be altered with regard to rates and routes of internalization.
With regard to toxicity and neurodegeneration, the signaling pathways that are linked to PrPC could likewise be modified by alterations in the lipid composition of the GPI-anchor as well as by proteolysis of PrPC. These issues will be briefly discussed in the following sections.
Signaling Through PrPC
Many reviews are dedicated to the role of signaling through PrPC in cell adhesion and in neurite extension/maintenance.Citation73,Citation74 Here we will only focus on how alterations of lipid rafts or changes in the GPI-anchor could lead to disturbances in signaling implicated in prion diseases and, briefly, other proteinopathies.
As pointed out above, in vitro-studies from the group of Bate and Williams show that manipulation of the GPI-anchor composition leads to modulation of prion infection, implicating phospholipase A2 (cPLA2) as the toxic signaling pathway.Citation75,Citation76 cPLA2 is involved in the generation of signaling molecules through the arachidonic acid pathway but also in membrane trafficking (for a review see ref.Citation77). On the one hand, clustering of PrPC, either mediated through antibodies or by addition of PrPSc, increases the cholesterol content in membranes and activates cPLA2 signaling, which then triggers synaptic damage. cPLA2 activation and synaptic damage could not be observed after clustering of Thy-1, indicating a specific effect of PrPC. The authors inferred that it is the clustering of the GPI-anchors, either caused by antibody cross-linking or PrPSc aggregation, that leads to neurotoxic effects. Importantly, when either the sialic acid or (one or both) acyl-chains are removed from the GPI-anchor of PrPC, or when the acylation of the GPI-anchor of PrPSc is altered, these neurotoxic effects are not observed. The authors proposed that the membrane composition is altered in the presence of either monoacylated or desialylated PrPC and that cPLA2 can consequently not be activated.Citation78A similar scenario is observed when PrPC is administered to prion-infected neuronal cell lines and integrates itself into the outer leaflet of the plasma membrane. This leads to increased amounts of cholesterol content in membranes and increased activation of cPLA2, together with an increased production of PrPSc. On the contrary, when monoacylated PrPC (PrPC-G-lyso-PI) is introduced, these effects are abolished. PrPC-G-lyso-PI is not present in lipid rafts, is not converted to PrPSc, and seems to displace cPLA2 from PrPSc-containing rafts, thus no longer permitting toxic signaling. Moreover, a decrease in PrPSc production is observed suggesting either that PrPC-G-lyso-PI is an inefficient template for conversion or that it competes with PrPC for partners involved in endocytosis, thus altering the trafficking and limiting the interactions between PrPC and PrPSc.Citation79,Citation80
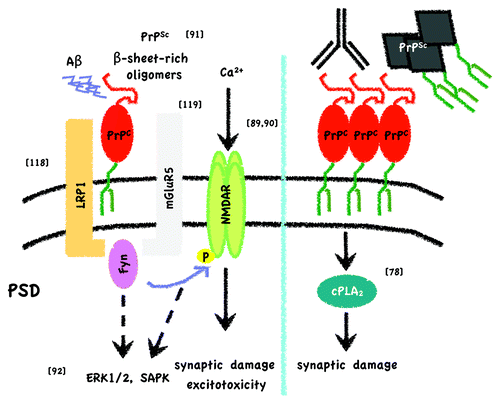
Other partners for signal transduction via PrPC have been described. For instance, PrPC has been involved in the inhibition of excitotoxicity mediated by the N-methyl-D-aspartate receptor (NMDAR).Citation81 It has been hypothesized that this regulation is executed either through direct interaction of PrPC with NMDAR subunits (PrPC co-immunoprecipitates with the NR2D subunit of the NMDAR) or via protein kinasesCitation82 such as the Src tyrosine kinase family member Fyn.Citation83 Of note, NMDARs localize in lipid raft domains but can also be present in non-raft domains after phosphorylation.Citation84 Moreover, cholesterol depletion reduces localization of NMDARs in lipid rafts and leads to neuroprotection.Citation85 On the other hand, Mouillet-Richard et al. first described Fyn as being activated by crosslinking of PrPC.Citation86 Since then, this pathway has been implicated in many of the functions that are attributed to PrPC, for instance its role as a neuronal receptor for toxic amyloid β (Aβ) oligomers.Citation87 Binding of Aβ oligomers derived from patients with Alzheimer disease activates the Fyn pathway leading to NR2B phosphorylation and altered NMDAR localization.Citation88-Citation90 However, clustering of PrPC by crosslinking failed to activate this pathway, suggesting that binding of β-sheet-rich oligomers leads to a conformational change in PrPC that is necessary to activate this pathway. It has been suggested that the concept of PrPC as a receptor for toxic oligomers can be generalized for other β-sheet-rich oligomers found in different neurodegenerative conditions (including PrPSc in prion diseases) and that the toxicity following this interaction is mediated by NMDAR.Citation91 The same authors highlighted the importance of PrPC localization in lipid rafts to mediate toxic signaling, since a transmembrane form of PrP (CD4PrP) did not mediate any toxic effects. Other kinases that are altered as a consequence of PrPSc binding to PrPC include ERK1/2, JNK, and the CREB transcription factor.Citation92,Citation93 A schematic draw synthesizing the issues discussed here is shown in .
Figure 2. Simplified scheme of toxic signaling through PrPC. On the one hand (left) it has been described that PrPC can bind Aβ oligomers at the post-synaptic density (PSD), either derived from recombinant sources or from brains of Alzheimer disease patients, and elicit toxicity through Fyn activation, NMDAR phosphorylation and altered localization.Citation88,Citation90 Phosphorylation of NMDAR subunits would then lead to dendritic spine loss and excitotoxicty. As long as PrPC is attached to the outer leaflet of the membrane and Fyn to the inner, it has been proposed that a transmembrane protein should act as a scaffold. LRP1Citation118 and mGluR5Citation119 have been suggested as the interacting transmembrane partners after Aβ oligomer binding to PrPC. Resenberger et al. also showed that this signaling cascade can apply for various β-sheet rich conformers.Citation91 In the case of PrPSc binding, apart from Fyn activation, other signaling targets, such as ERK1/2, p38 and JNK, are seen to be activated in neuronal cell lines.Citation92 In addition, it might be hypothesized that β-sheet rich oligomer-associated NMDAR activation per se induces MAP kinase pathways. On the other hand (right) experiments by Bate and Williams demonstrated that after either crosslinking of PrPC with antibodies or by applying PrPSc to cortical primary neurons, there is an increase in membrane cholesterol content that leads to a toxic pathway implicating cPLA2 and arachidonic acid metabolites.Citation78 Reference to original studies is given by the numbers in brackets.
Anchorage and Localization of PrPC May Influence its Proteolytic Processing
Proteolytic processing reflects an additional aspect of prion protein biology that is likely to be influenced by modifications of the membrane lipid composition or in the GPI-anchor attachment resulting in alterations in PrPC localization. In recent years it has become increasingly obvious that evolutionary conserved cleavage events, especially the α-cleavage in the center of PrPCCitation94 and the shedding in close proximity to its GPI-anchor,Citation95,Citation96 not only regulate PrPC membrane homeostasis and reduce amounts of full-length PrPC and, thus, the mediator of neurotoxic signaling and the substrate for conformational prion conversion. In addition, these cleavages produce biologically active fragmentsCitation97,Citation98 (reviewed in ref. Citation99). Of note, the released soluble fragments, N1 (for α-cleavage) and shed PrP as well as their artificial correlates, have recently been linked with neuroprotection by blocking both the access of toxic oligomeric protein species to the cell as well as the conversion process in the context of prion diseases.Citation91,Citation100-Citation107 Thus, it is not surprising that these cleavage events are currently discussed as therapeutic targets with regard to prion diseases and other neurodegenerative proteinopathies.
The identity of the protease(s) responsible for the α-cleavage remains a matter of controversy (reviewed in refs. Citation108 and Citation109) and a recent study suggests a more complex scenario than initially expected, involving different members of the ADAM family of metalloproteases.Citation110 In contrast, the role of ADAM10 as the major PrPC sheddase has been established by recent studies.Citation95,Citation96,Citation107,Citation111
While knowledge on the concrete localization of the two cleavage events is limitedCitation54,Citation111-Citation113 both processing steps seem to depend on PrPC membrane attachment. As a matter of implicitness, for a protein to be proteolytically cleaved it has to meet and interact with the relevant protease. Therefore, it would not be surprising if processes modifying the GPI-anchor attachment of PrPC, resulting in altered structure or sorting/localization, or disturbing the integrity of lipid rafts may influence the substrate-protease interaction and alter the neuroprotective processing of PrPC. In fact, it has been shown for other ADAM10 substrates mainly present in lipid rafts, that depletion of these microdomains significantly increased the proteolytic processing due to increased interaction with the protease (as active ADAM10 is preferentially found outside of rafts).Citation114-Citation117 Moreover, attachment of a GPI-anchor to ADAM10 was shown to target the protease to lipid rafts, which resulted in altered processing of the amyloid precursor protein.Citation116 With regard to PrPC more research is necessary to clarify whether manipulations of this kind might offer a treatment option for prion diseases and other proteinopathies.
Concluding Remarks
Increasing evidence locates PrPC as a key player in mediating neurotoxic signaling not only in prion diseases but in other neurodegenerative diseases such as Alzheimer disease. Understanding the importance of membrane localization of PrPC in lipid rafts as well as how the anchorage of PrPC can influence disease progression is of outstanding importance in order to develop further therapeutic strategies.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
This work is supported by grants of the Werner-Otto-Stiftung (to H.A.) and the Deutsche Forschungsgemeinschaft (DFG, GRK1459 and FOR885).
Related Research Data
References
- Rudd PM, Merry AH, Wormald MR, Dwek RA. Glycosylation and prion protein. CurrOpinStructBiol 2002; 12:578 - 86; http://dx.doi.org/10.1016/S0959-440X(02)00377-9; PMID: 12464308
- Stahl N, Baldwin MA, Teplow DB, Hood L, Gibson BW, Burlingame AL, Prusiner SB. Structural studies of the scrapie prion protein using mass spectrometry and amino acid sequencing. Biochemistry 1993; 32:1991 - 2002; http://dx.doi.org/10.1021/bi00059a016; PMID: 8448158
- Prusiner SB. Prions. ProcNatlAcadSci U S A 1998; 95:13363 - 83; http://dx.doi.org/10.1073/pnas.95.23.13363; PMID: 9811807
- Fujita M, Kinoshita T. Structural remodeling of GPI anchors during biosynthesis and after attachment to proteins. FEBS Lett 2010; 584:1670 - 7; http://dx.doi.org/10.1016/j.febslet.2009.10.079; PMID: 19883648
- FergusonMAJ, KinoshitaT, HartGW. Glycosylphosphatidylinositol Anchors. 2nd ed. Cold Spring Harbor: Cold Spring Harbor Laboratory Press; 2009. Chapter 11
- Paulick MG, Bertozzi CR. The glycosylphosphatidylinositol anchor: a complex membrane-anchoring structure for proteins. Biochemistry 2008; 47:6991 - 7000; http://dx.doi.org/10.1021/bi8006324; PMID: 18557633
- Orlean P, Menon AK. Thematic review series: lipid posttranslational modifications. GPI anchoring of protein in yeast and mammalian cells, or: how we learned to stop worrying and love glycophospholipids. J Lipid Res 2007; 48:993 - 1011; http://dx.doi.org/10.1194/jlr.R700002-JLR200; PMID: 17361015
- Morandat S, Bortolato M, Roux B. Role of GPI-anchored enzyme in liposome detergent-resistance. J MembrBiol 2003; 191:215 - 21; http://dx.doi.org/10.1007/s00232-002-1056-y; PMID: 12571756
- Barboni E, Rivero BP, George AJ, Martin SR, Renoup DV, Hounsell EF, Barber PC, Morris RJ. The glycophosphatidylinositol anchor affects the conformation of Thy-1 protein. J Cell Sci 1995; 108:487 - 97; PMID: 7539435
- Kukulansky T, Abramovitch S, Hollander N. Cleavage of the glycosylphosphatidylinositol anchor affects the reactivity of thy-1 with antibodies. J Immunol 1999; 162:5993 - 7; PMID: 10229838
- Legler DF, Doucey MA, Schneider P, Chapatte L, Bender FC, Bron C. Differential insertion of GPI-anchored GFPs into lipid rafts of live cells. FASEB J 2005; 19:73 - 5; PMID: 15516372
- Medof ME, Nagarajan S, Tykocinski ML. Cell-surface engineering with GPI-anchored proteins. FASEB J 1996; 10:574 - 86; PMID: 8621057
- Liu T, Li R, Pan T, Liu D, Petersen RB, Wong BS, Gambetti P, Sy MS. Intercellular transfer of the cellular prion protein. J BiolChem 2002; 277:47671 - 8; http://dx.doi.org/10.1074/jbc.M207458200; PMID: 12359724
- Vidugiriene J, Menon AK. The GPI anchor of cell-surface proteins is synthesized on the cytoplasmic face of the endoplasmic reticulum. J Cell Biol 1994; 127:333 - 41; http://dx.doi.org/10.1083/jcb.127.2.333; PMID: 7929579
- Brewis IA, Ferguson MA, Mehlert A, Turner AJ, Hooper NM. Structures of the glycosyl-phosphatidylinositol anchors of porcine and human renal membrane dipeptidase. Comprehensive structural studies on the porcine anchor and interspecies comparison of the glycan core structures. J BiolChem 1995; 270:22946 - 56; http://dx.doi.org/10.1074/jbc.270.39.22946; PMID: 7559431
- Meri S, Lehto T, Sutton CW, Tyynelä J, Baumann M. Structural composition and functional characterization of soluble CD59: heterogeneity of the oligosaccharide and glycophosphoinositol (GPI) anchor revealed by laser-desorption mass spectrometric analysis. Biochem J 1996; 316:923 - 35; PMID: 8670172
- Stahl N, Baldwin MA, Burlingame AL, Prusiner SB. Identification of glycoinositol phospholipid linked and truncated forms of the scrapie prion protein. Biochemistry 1990; 29:8879 - 84; http://dx.doi.org/10.1021/bi00490a001; PMID: 1980209
- Baldwin MA. Analysis of glycosylphosphatidylinositol protein anchors: the prion protein. Methods Enzymol 2005; 405:172 - 87; http://dx.doi.org/10.1016/S0076-6879(05)05008-1; PMID: 16413315
- Simons K, Sampaio JL. Membrane organization and lipid rafts. Cold Spring HarbPerspectBiol 2011; 3:a004697; http://dx.doi.org/10.1101/cshperspect.a004697; PMID: 21628426
- Udenfriend S, Kodukula K. How glycosylphosphatidylinositol-anchored membrane proteins are made. Annu Rev Biochem 1995; 64:563 - 91; http://dx.doi.org/10.1146/annurev.bi.64.070195.003023; PMID: 7574493
- Fujita M, Jigami Y. Lipid remodeling of GPI-anchored proteins and its function. BiochimBiophysActa 2008; 1780:410 - 20; http://dx.doi.org/10.1016/j.bbagen.2007.08.009; PMID: 17913366
- Lingwood D, Simons K. Lipid rafts as a membrane-organizing principle. Science 2010; 327:46 - 50; http://dx.doi.org/10.1126/science.1174621; PMID: 20044567
- Tsui-Pierchala BA, Encinas M, Milbrandt J, Johnson EM Jr.. Lipid rafts in neuronal signaling and function. Trends Neurosci 2002; 25:412 - 7; http://dx.doi.org/10.1016/S0166-2236(02)02215-4; PMID: 12127758
- Stefanová I, Horejsí V, Ansotegui IJ, Knapp W, Stockinger H. GPI-anchored cell-surface molecules complexed to protein tyrosine kinases. Science 1991; 254:1016 - 9; http://dx.doi.org/10.1126/science.1719635; PMID: 1719635
- Chen Y, Veracini L, Benistant C, Jacobson K. The transmembrane protein CBP plays a role in transiently anchoring small clusters of Thy-1, a GPI-anchored protein, to the cytoskeleton. J Cell Sci 2009; 122:3966 - 72; http://dx.doi.org/10.1242/jcs.049346; PMID: 19825940
- de Hoop MJ, Dotti CG. Membrane traffic in polarized neurons in culture. J Cell SciSuppl 1993; 17:85 - 92; http://dx.doi.org/10.1242/jcs.1993.Supplement_17.13; PMID: 8144707
- van Meer G, Simons K. Lipid polarity and sorting in epithelial cells. J Cell Biochem 1988; 36:51 - 8; http://dx.doi.org/10.1002/jcb.240360106; PMID: 3277985
- Brown DA, Crise B, Rose JK. Mechanism of membrane anchoring affects polarized expression of two proteins in MDCK cells. Science 1989; 245:1499 - 501; http://dx.doi.org/10.1126/science.2571189; PMID: 2571189
- Lisanti MP, Caras IW, Davitz MA, Rodriguez-Boulan E. A glycophospholipid membrane anchor acts as an apical targeting signal in polarized epithelial cells. J Cell Biol 1989; 109:2145 - 56; http://dx.doi.org/10.1083/jcb.109.5.2145; PMID: 2478564
- Sarnataro D, Paladino S, Campana V, Grassi J, Nitsch L, Zurzolo C. PrPC is sorted to the basolateral membrane of epithelial cells independently of its association with rafts. Traffic 2002; 3:810 - 21; http://dx.doi.org/10.1034/j.1600-0854.2002.31106.x; PMID: 12383347
- Paladino S, Sarnataro D, Pillich R, Tivodar S, Nitsch L, Zurzolo C. Protein oligomerization modulates raft partitioning and apical sorting of GPI-anchored proteins. J Cell Biol 2004; 167:699 - 709; http://dx.doi.org/10.1083/jcb.200407094; PMID: 15557121
- Paladino S, Lebreton S, Tivodar S, Campana V, Tempre R, Zurzolo C. Different GPI-attachment signals affect the oligomerisation of GPI-anchored proteins and their apical sorting. J Cell Sci 2008; 121:4001 - 7; http://dx.doi.org/10.1242/jcs.036038; PMID: 19056670
- Puig B, Altmeppen HC, Thurm D, Geissen M, Conrad C, Braulke T, Glatzel M. N-glycans and glycosylphosphatidylinositol-anchor act on polarized sorting of mouse PrP(C) in Madin-Darby canine kidney cells. PLoS One 2011; 6:e24624; http://dx.doi.org/10.1371/journal.pone.0024624; PMID: 21931781
- Madore N, Smith KL, Graham CH, Jen A, Brady K, Hall S, Morris R. Functionally different GPI proteins are organized in different domains on the neuronal surface. EMBO J 1999; 18:6917 - 26; http://dx.doi.org/10.1093/emboj/18.24.6917; PMID: 10601014
- Brügger B, Graham C, Leibrecht I, Mombelli E, Jen A, Wieland F, Morris R. The membrane domains occupied by glycosylphosphatidylinositol-anchored prion protein and Thy-1 differ in lipid composition. J BiolChem 2004; 279:7530 - 6; http://dx.doi.org/10.1074/jbc.M310207200; PMID: 14660659
- Tivodar S, Paladino S, Pillich R, Prinetti A, Chigorno V, van Meer G, Sonnino S, Zurzolo C. Analysis of detergent-resistant membranes associated with apical and basolateral GPI-anchored proteins in polarized epithelial cells. FEBS Lett 2006; 580:5705 - 12; http://dx.doi.org/10.1016/j.febslet.2006.09.022; PMID: 17007841
- Mironov A Jr., Latawiec D, Wille H, Bouzamondo-Bernstein E, Legname G, Williamson RA, Burton D, DeArmond SJ, Prusiner SB, Peters PJ. Cytosolic prion protein in neurons. J Neurosci 2003; 23:7183 - 93; PMID: 12904479
- Galvan C, Camoletto PG, Dotti CG, Aguzzi A, Ledesma MD. Proper axonal distribution of PrP(C) depends on cholesterol-sphingomyelin-enriched membrane domains and is developmentally regulated in hippocampal neurons. Mol Cell Neurosci 2005; 30:304 - 15; http://dx.doi.org/10.1016/j.mcn.2005.07.003; PMID: 16139509
- Chen R, Knez JJ, Merrick WC, Medof ME. Comparative efficiencies of C-terminal signals of native glycophosphatidylinositol (GPI)-anchored proproteins in conferring GPI-anchoring. J Cell Biochem 2001; 84:68 - 83; http://dx.doi.org/10.1002/jcb.1267; PMID: 11746517
- Nicholson TB, Stanners CP. Specific inhibition of GPI-anchored protein function by homing and self-association of specific GPI anchors. J Cell Biol 2006; 175:647 - 59; http://dx.doi.org/10.1083/jcb.200605001; PMID: 17101695
- Nicholson TB, Stanners CP. Identification of a novel functional specificity signal within the GPI anchor signal sequence of carcinoembryonic antigen. J Cell Biol 2007; 177:211 - 8; http://dx.doi.org/10.1083/jcb.200701158; PMID: 17438079
- Wadsworth JD, Hill AF, Beck JA, Collinge J. Molecular and clinical classification of human prion disease. Br Med Bull 2003; 66:241 - 54; http://dx.doi.org/10.1093/bmb/66.1.241; PMID: 14522862
- Gu Y, Singh A, Bose S, Singh N. Pathogenic mutations in the glycosylphosphatidylinositol signal peptide of PrP modulate its topology in neuroblastoma cells. Mol Cell Neurosci 2008; 37:647 - 56; http://dx.doi.org/10.1016/j.mcn.2007.08.018; PMID: 18325785
- Guizzunti G, Zurzolo C. The fate of PrP GPI-anchor signal peptide is modulated by P238S pathogenic mutation. Traffic 2014; 15:78 - 93; http://dx.doi.org/10.1111/tra.12126; PMID: 24112521
- Borchelt DR, Taraboulos A, Prusiner SB. Evidence for synthesis of scrapie prion proteins in the endocytic pathway. J BiolChem 1992; 267:16188 - 99; PMID: 1353761
- Béranger F, Mangé A, Goud B, Lehmann S. Stimulation of PrP(C) retrograde transport toward the endoplasmic reticulum increases accumulation of PrP(Sc) in prion-infected cells. J BiolChem 2002; 277:38972 - 7; http://dx.doi.org/10.1074/jbc.M205110200; PMID: 12163492
- Marijanovic Z, Caputo A, Campana V, Zurzolo C. Identification of an intracellular site of prion conversion. PLoSPathog 2009; 5:e1000426; http://dx.doi.org/10.1371/journal.ppat.1000426; PMID: 19424437
- Baron GS, Wehrly K, Dorward DW, Chesebro B, Caughey B. Conversion of raft associated prion protein to the protease-resistant state requires insertion of PrP-res (PrP(Sc)) into contiguous membranes. EMBO J 2002; 21:1031 - 40; http://dx.doi.org/10.1093/emboj/21.5.1031; PMID: 11867531
- Goold R, Rabbanian S, Sutton L, Andre R, Arora P, Moonga J, Clarke AR, Schiavo G, Jat P, Collinge J, et al. Rapid cell-surface prion protein conversion revealed using a novel cell system. Nat Commun 2011; 2:281; http://dx.doi.org/10.1038/ncomms1282; PMID: 21505437
- Goold R, McKinnon C, Rabbanian S, Collinge J, Schiavo G, Tabrizi SJ. Alternative fates of newly formed PrPSc upon prion conversion on the plasma membrane. J Cell Sci 2013; 126:3552 - 62; http://dx.doi.org/10.1242/jcs.120477; PMID: 23813960
- Taraboulos A, Scott M, Semenov A, Avrahami D, Laszlo L, Prusiner SB. Cholesterol depletion and modification of COOH-terminal targeting sequence of the prion protein inhibit formation of the scrapie isoform. J Cell Biol 1995; 129:121 - 32; http://dx.doi.org/10.1083/jcb.129.1.121; PMID: 7698979
- Mangé A, Nishida N, Milhavet O, McMahon HE, Casanova D, Lehmann S. Amphotericin B inhibits the generation of the scrapie isoform of the prion protein in infected cultures. J Virol 2000; 74:3135 - 40; http://dx.doi.org/10.1128/JVI.74.7.3135-3140.2000; PMID: 10708429
- Marella M, Lehmann S, Grassi J, Chabry J. Filipin prevents pathological prion protein accumulation by reducing endocytosis and inducing cellular PrP release. J BiolChem 2002; 277:25457 - 64; http://dx.doi.org/10.1074/jbc.M203248200; PMID: 11994310
- Taraboulos A, Scott M, Semenov A, Avrahami D, Laszlo L, Prusiner SB. Cholesterol depletion and modification of COOH-terminal targeting sequence of the prion protein inhibit formation of the scrapie isoform. J Cell Biol 1995; 129:121 - 32; http://dx.doi.org/10.1083/jcb.129.1.121; PMID: 7698979
- Kocisko DA, Come JH, Priola SA, Chesebro B, Raymond GJ, Lansbury PT, Caughey B. Cell-free formation of protease-resistant prion protein. Nature 1994; 370:471 - 4; http://dx.doi.org/10.1038/370471a0; PMID: 7913989
- McNally KL, Ward AE, Priola SA. Cells expressing anchorless prion protein are resistant to scrapie infection. J Virol 2009; 83:4469 - 75; http://dx.doi.org/10.1128/JVI.02412-08; PMID: 19225008
- Priola SA, McNally KL. The role of the prion protein membrane anchor in prion infection. Prion 2009; 3:134 - 8; http://dx.doi.org/10.4161/pri.3.3.9771; PMID: 19786843
- Speare JO, Offerdahl DK, Hasenkrug A, Carmody AB, Baron GS. GPI anchoring facilitates propagation and spread of misfolded Sup35 aggregates in mammalian cells. EMBO J 2010; 29:782 - 94; http://dx.doi.org/10.1038/emboj.2009.392; PMID: 20057357
- Fernández-Borges N, Chianini F, Eraña H, Vidal E, Eaton SL, Pintado B, Finlayson J, Dagleish MP, Castilla J. Naturally prion resistant mammals: a utopia?. Prion 2012; 6:425 - 9; http://dx.doi.org/10.4161/pri.22057; PMID: 22954650
- Nisbet RM, Harrison CF, Lawson VA, Masters CL, Cappai R, Hill AF. Residues surrounding the glycosylphosphatidylinositol anchor attachment site of PrP modulate prion infection: insight from the resistance of rabbits to prion disease. J Virol 2010; 84:6678 - 86; http://dx.doi.org/10.1128/JVI.02709-09; PMID: 20427543
- Jeffrey M, McGovern G, Sisó S, González L. Cellular and sub-cellular pathology of animal prion diseases: relationship between morphological changes, accumulation of abnormal prion protein and clinical disease. ActaNeuropathol 2011; 121:113 - 34; http://dx.doi.org/10.1007/s00401-010-0700-3; PMID: 20532540
- Büeler H, Aguzzi A, Sailer A, Greiner RA, Autenried P, Aguet M, Weissmann C. Mice devoid of PrP are resistant to scrapie. Cell 1993; 73:1339 - 47; http://dx.doi.org/10.1016/0092-8674(93)90360-3; PMID: 8100741
- Mallucci G, Dickinson A, Linehan J, Klöhn PC, Brandner S, Collinge J. Depleting neuronal PrP in prion infection prevents disease and reverses spongiosis. Science 2003; 302:871 - 4; http://dx.doi.org/10.1126/science.1090187; PMID: 14593181
- Sandberg MK, Al-Doujaily H, Sharps B, Clarke AR, Collinge J. Prion propagation and toxicity in vivo occur in two distinct mechanistic phases. Nature 2011; 470:540 - 2; http://dx.doi.org/10.1038/nature09768; PMID: 21350487
- Chesebro B, Race B, Meade-White K, Lacasse R, Race R, Klingeborn M, Striebel J, Dorward D, McGovern G, Jeffrey M. Fatal transmissible amyloid encephalopathy: a new type of prion disease associated with lack of prion protein membrane anchoring. PLoSPathog 2010; 6:e1000800; http://dx.doi.org/10.1371/journal.ppat.1000800; PMID: 20221436
- Fasano C, Campana V, Zurzolo C. Prions: protein only or something more? Overview of potential prion cofactors. J MolNeurosci 2006; 29:195 - 214; http://dx.doi.org/10.1385/JMN:29:3:195; PMID: 17085779
- Taylor DR, Whitehouse IJ, Hooper NM. Glypican-1 mediates both prion protein lipid raft association and disease isoform formation. PLoSPathog 2009; 5:e1000666; http://dx.doi.org/10.1371/journal.ppat.1000666; PMID: 19936054
- Fivaz M, Vilbois F, Thurnheer S, Pasquali C, Abrami L, Bickel PE, Parton RG, van der Goot FG. Differential sorting and fate of endocytosed GPI-anchored proteins. EMBO J 2002; 21:3989 - 4000; http://dx.doi.org/10.1093/emboj/cdf398; PMID: 12145200
- Sunyach C, Jen A, Deng J, Fitzgerald KT, Frobert Y, Grassi J, McCaffrey MW, Morris R. The mechanism of internalization of glycosylphosphatidylinositol-anchored prion protein. EMBO J 2003; 22:3591 - 601; http://dx.doi.org/10.1093/emboj/cdg344; PMID: 12853474
- Taylor DR, Hooper NM. The low-density lipoprotein receptor-related protein 1 (LRP1) mediates the endocytosis of the cellular prion protein. Biochem J 2007; 402:17 - 23; http://dx.doi.org/10.1042/BJ20061736; PMID: 17155929
- Parkyn CJ, Vermeulen EG, Mootoosamy RC, Sunyach C, Jacobsen C, Oxvig C, Moestrup S, Liu Q, Bu G, Jen A, et al. LRP1 controls biosynthetic and endocytic trafficking of neuronal prion protein. J Cell Sci 2008; 121:773 - 83; http://dx.doi.org/10.1242/jcs.021816; PMID: 18285446
- Sarnataro D, Caputo A, Casanova P, Puri C, Paladino S, Tivodar SS, Campana V, Tacchetti C, Zurzolo C. Lipid rafts and clathrin cooperate in the internalization of PrP in epithelial FRT cells. PLoS One 2009; 4:e5829; http://dx.doi.org/10.1371/journal.pone.0005829; PMID: 19503793
- Linden R, Martins VR, Prado MA, Cammarota M, Izquierdo I, Brentani RR. Physiology of the prion protein. Physiol Rev 2008; 88:673 - 728; http://dx.doi.org/10.1152/physrev.00007.2007; PMID: 18391177
- Biasini E, Turnbaugh JA, Unterberger U, Harris DA. Prion protein at the crossroads of physiology and disease. Trends Neurosci 2012; 35:92 - 103; http://dx.doi.org/10.1016/j.tins.2011.10.002; PMID: 22137337
- Bate C, Williams A. Role of glycosylphosphatidylinositols in the activation of phospholipase A2 and the neurotoxicity of prions. J Gen Virol 2004; 85:3797 - 804; http://dx.doi.org/10.1099/vir.0.80366-0; PMID: 15557253
- Bate C, Tayebi M, Williams A. Phospholipase A2 inhibitors protect against prion and Abeta mediated synapse degeneration. MolNeurodegener 2010; 5:13; http://dx.doi.org/10.1186/1750-1326-5-13; PMID: 20374666
- Brown WJ, Chambers K, Doody A. Phospholipase A2 (PLA2) enzymes in membrane trafficking: mediators of membrane shape and function. Traffic 2003; 4:214 - 21; http://dx.doi.org/10.1034/j.1600-0854.2003.00078.x; PMID: 12694560
- Bate C, Williams A. Neurodegeneration induced by clustering of sialylatedglycosylphosphatidylinositols of prion proteins. J BiolChem 2012; 287:7935 - 44; http://dx.doi.org/10.1074/jbc.M111.275743; PMID: 22262833
- Bate C, Williams A. Monoacylated cellular prion protein modifies cell membranes, inhibits cell signaling, and reduces prion formation. J BiolChem 2011; 286:8752 - 8; http://dx.doi.org/10.1074/jbc.M110.186833; PMID: 21212283
- Bate C, Williams A. The cellular prion protein with a monoacylatedglycosylphosphatidylinositol anchor modifies cell membranes, inhibits cell signaling and reduces prion formation. Prion 2011; 5:65 - 8; http://dx.doi.org/10.4161/pri.5.2.16095; PMID: 21738009
- Khosravani H, Zhang Y, Tsutsui S, Hameed S, Altier C, Hamid J, Chen L, Villemaire M, Ali Z, Jirik FR, et al. Prion protein attenuates excitotoxicity by inhibiting NMDA receptors. J Cell Biol 2008; 181:551 - 65; http://dx.doi.org/10.1083/jcb.200711002; PMID: 18443219
- Wang YT, Salter MW. Regulation of NMDA receptors by tyrosine kinases and phosphatases. Nature 1994; 369:233 - 5; http://dx.doi.org/10.1038/369233a0; PMID: 7514272
- Salter MW, Kalia LV. Src kinases: a hub for NMDA receptor regulation. Nat Rev Neurosci 2004; 5:317 - 28; http://dx.doi.org/10.1038/nrn1368; PMID: 15034556
- Swanwick CC, Shapiro ME, Yi Z, Chang K, Wenthold RJ. NMDA receptors interact with flotillin-1 and -2, lipid raft-associated proteins. FEBS Lett 2009; 583:1226 - 30; http://dx.doi.org/10.1016/j.febslet.2009.03.017; PMID: 19298817
- Ponce J, de la Ossa NP, Hurtado O, Millan M, Arenillas JF, Dávalos A, Gasull T. Simvastatin reduces the association of NMDA receptors to lipid rafts: a cholesterol-mediated effect in neuroprotection. Stroke 2008; 39:1269 - 75; http://dx.doi.org/10.1161/STROKEAHA.107.498923; PMID: 18323503
- Mouillet-Richard S, Ermonval M, Chebassier C, Laplanche JL, Lehmann S, Launay JM, Kellermann O. Signal transduction through prion protein. Science 2000; 289:1925 - 8; http://dx.doi.org/10.1126/science.289.5486.1925; PMID: 10988071
- Laurén J, Gimbel DA, Nygaard HB, Gilbert JW, Strittmatter SM. Cellular prion protein mediates impairment of synaptic plasticity by amyloid-beta oligomers. Nature 2009; 457:1128 - 32; http://dx.doi.org/10.1038/nature07761; PMID: 19242475
- Um JW, Nygaard HB, Heiss JK, Kostylev MA, Stagi M, Vortmeyer A, Wisniewski T, Gunther EC, Strittmatter SM. Alzheimer amyloid-β oligomer bound to postsynaptic prion protein activates Fyn to impair neurons. Nat Neurosci 2012; 15:1227 - 35; http://dx.doi.org/10.1038/nn.3178; PMID: 22820466
- Um JW, Strittmatter SM. Amyloid-β induced signaling by cellular prion protein and Fyn kinase in Alzheimer disease. Prion 2013; 7:37 - 41; http://dx.doi.org/10.4161/pri.22212; PMID: 22987042
- Larson ME, Lesné SE. Soluble Aβ oligomer production and toxicity. J Neurochem 2012; 120:Suppl 1 125 - 39; http://dx.doi.org/10.1111/j.1471-4159.2011.07478.x; PMID: 22121920
- Resenberger UK, Harmeier A, Woerner AC, Goodman JL, Müller V, Krishnan R, Vabulas RM, Kretzschmar HA, Lindquist S, Hartl FU, et al. The cellular prion protein mediates neurotoxic signalling of β-sheet-rich conformers independent of prion replication. EMBO J 2011; 30:2057 - 70; http://dx.doi.org/10.1038/emboj.2011.86; PMID: 21441896
- Pradines E, Hernandez-Rapp J, Villa-Diaz A, Dakowski C, Ardila-Osorio H, Haik S, Schneider B, Launay JM, Kellermann O, Torres JM, et al. Pathogenic prions deviate PrP(C) signaling in neuronal cells and impair A-beta clearance. Cell Death Dis 2013; 4:e456; http://dx.doi.org/10.1038/cddis.2012.195; PMID: 23303130
- Rambold AS, Müller V, Ron U, Ben-Tal N, Winklhofer KF, Tatzelt J. Stress-protective signalling of prion protein is corrupted by scrapie prions. EMBO J 2008; 27:1974 - 84; http://dx.doi.org/10.1038/emboj.2008.122; PMID: 18566584
- Chen SG, Teplow DB, Parchi P, Teller JK, Gambetti P, Autilio-Gambetti L. Truncated forms of the human prion protein in normal brain and in prion diseases. J BiolChem 1995; 270:19173 - 80; http://dx.doi.org/10.1074/jbc.270.32.19173; PMID: 7642585
- Taylor DR, Parkin ET, Cocklin SL, Ault JR, Ashcroft AE, Turner AJ, Hooper NM. Role of ADAMs in the ectodomain shedding and conformational conversion of the prion protein. J BiolChem 2009; 284:22590 - 600; http://dx.doi.org/10.1074/jbc.M109.032599; PMID: 19564338
- Altmeppen HC, Prox J, Puig B, Kluth MA, Bernreuther C, Thurm D, Jorissen E, Petrowitz B, Bartsch U, De Strooper B, et al. Lack of a-disintegrin-and-metalloproteinase ADAM10 leads to intracellular accumulation and loss of shedding of the cellular prion protein in vivo. MolNeurodegener 2011; 6:36; http://dx.doi.org/10.1186/1750-1326-6-36; PMID: 21619641
- Sunyach C, Cisse MA, da Costa CA, Vincent B, Checler F. The C-terminal products of cellular prion protein processing, C1 and C2, exert distinct influence on p53-dependent staurosporine-induced caspase-3 activation. J BiolChem 2007; 282:1956 - 63; http://dx.doi.org/10.1074/jbc.M609663200; PMID: 17121821
- Westergard L, Turnbaugh JA, Harris DA. A naturally occurring C-terminal fragment of the prion protein (PrP) delays disease and acts as a dominant-negative inhibitor of PrPSc formation. J BiolChem 2011; 286:44234 - 42; http://dx.doi.org/10.1074/jbc.M111.286195; PMID: 22025612
- Altmeppen HC, Prox J, Puig B, Dohler F, Falker C, Krasemann S, Glatzel M. Roles of endoproteolytic α-cleavage and shedding of the prion protein in neurodegeneration. FEBS J 2013; 280:4338 - 47; http://dx.doi.org/10.1111/febs.12196; PMID: 23413979
- Meier P, Genoud N, Prinz M, Maissen M, Rülicke T, Zurbriggen A, Raeber AJ, Aguzzi A. Soluble dimeric prion protein binds PrP(Sc) in vivo and antagonizes prion disease. Cell 2003; 113:49 - 60; http://dx.doi.org/10.1016/S0092-8674(03)00201-0; PMID: 12679034
- Guillot-Sestier MV, Sunyach C, Druon C, Scarzello S, Checler F. The alpha-secretase-derived N-terminal product of cellular prion, N1, displays neuroprotective function in vitro and in vivo. J BiolChem 2009; 284:35973 - 86; http://dx.doi.org/10.1074/jbc.M109.051086; PMID: 19850936
- Guillot-Sestier MV, Checler F. α-Secretase-derived cleavage of cellular prion yields biologically active catabolites with distinct functions. Neurodegener Dis 2012; 10:294 - 7; http://dx.doi.org/10.1159/000333804; PMID: 22261541
- Calella AM, Farinelli M, Nuvolone M, Mirante O, Moos R, Falsig J, Mansuy IM, Aguzzi A. Prion protein and Abeta-related synaptic toxicity impairment. EMBO Mol Med 2010; 2:306 - 14; http://dx.doi.org/10.1002/emmm.201000082; PMID: 20665634
- Béland M, Motard J, Barbarin A, Roucou X. PrP(C) homodimerization stimulates the production of PrPC cleaved fragments PrPN1 and PrPC1. J Neurosci 2012; 32:13255 - 63; http://dx.doi.org/10.1523/JNEUROSCI.2236-12.2012; PMID: 22993441
- Nieznanski K, Choi JK, Chen S, Surewicz K, Surewicz WK. Soluble prion protein inhibits amyloid-β (Aβ) fibrillization and toxicity. J BiolChem 2012; 287:33104 - 8; http://dx.doi.org/10.1074/jbc.C112.400614; PMID: 22915585
- Fluharty BR, Biasini E, Stravalaci M, Sclip A, Diomede L, Balducci C, La Vitola P, Messa M, Colombo L, Forloni G, et al. An N-terminal fragment of the prion protein binds to amyloid-β oligomers and inhibits their neurotoxicity in vivo. J BiolChem 2013; 288:7857 - 66; http://dx.doi.org/10.1074/jbc.M112.423954; PMID: 23362282
- Ostapchenko VG, Beraldo FH, Guimarães AL, Mishra S, Guzman M, Fan J, Martins VR, Prado VF, Prado MA. Increased prion protein processing and expression of metabotropic glutamate receptor 1 in a mouse model of Alzheimer’s disease. J Neurochem 2013; 127:415 - 25; http://dx.doi.org/10.1111/jnc.12296; PMID: 23651058
- Altmeppen HC, Puig B, Dohler F, Thurm DK, Falker C, Krasemann S, Glatzel M. Proteolytic processing of the prion protein in health and disease. Am J Neurodegener Dis 2012; 1:15 - 31; PMID: 23383379
- Liang J, Kong Q. α-Cleavage of cellular prion protein. Prion 2012; 6:453 - 60; http://dx.doi.org/10.4161/pri.22511; PMID: 23052041
- McDonald AJ, Dibble JP, Evans EG, Millhauser GL. A New Paradigm for Enzymatic Control of α-Cleavage and β-Cleavage of the Prion Protein. J BiolChem 2014; 289:803 - 13; http://dx.doi.org/10.1074/jbc.M113.502351; PMID: 24247244
- Wik L, Klingeborn M, Willander H, Linne T. Separate mechanisms act concurrently to shed and release the prion protein from the cell. Prion 2012; 6:498 - 509; http://dx.doi.org/10.4161/pri.22588; PMID: 23093798
- Walmsley AR, Watt NT, Taylor DR, Perera WS, Hooper NM. alpha-cleavage of the prion protein occurs in a late compartment of the secretory pathway and is independent of lipid rafts. Mol Cell Neurosci 2009; 40:242 - 8; http://dx.doi.org/10.1016/j.mcn.2008.10.012; PMID: 19056496
- Liang J, Wang W, Sorensen D, Medina S, Ilchenko S, Kiselar J, Surewicz WK, Booth SA, Kong Q. Cellular prion protein regulates its own α-cleavage through ADAM8 in skeletal muscle. J BiolChem 2012; 287:16510 - 20; http://dx.doi.org/10.1074/jbc.M112.360891; PMID: 22447932
- Kojro E, Gimpl G, Lammich S, Marz W, Fahrenholz F. Low cholesterol stimulates the nonamyloidogenic pathway by its effect on the alpha -secretase ADAM 10. ProcNatlAcadSci U S A 2001; 98:5815 - 20; http://dx.doi.org/10.1073/pnas.081612998; PMID: 11309494
- Kojro E, Füger P, Prinzen C, Kanarek AM, Rat D, Endres K, Fahrenholz F, Postina R. Statins and the squalene synthase inhibitor zaragozic acid stimulate the non-amyloidogenic pathway of amyloid-beta protein precursor processing by suppression of cholesterol synthesis. J Alzheimers Dis 2010; 20:1215 - 31; PMID: 20413873
- Harris B, Pereira I, Parkin E. Targeting ADAM10 to lipid rafts in neuroblastoma SH-SY5Y cells impairs amyloidogenic processing of the amyloid precursor protein. Brain Res 2009; 1296:203 - 15; http://dx.doi.org/10.1016/j.brainres.2009.07.105; PMID: 19679113
- Murai T, Maruyama Y, Mio K, Nishiyama H, Suga M, Sato C. Low cholesterol triggers membrane microdomain-dependent CD44 shedding and suppresses tumor cell migration. J BiolChem 2011; 286:1999 - 2007; http://dx.doi.org/10.1074/jbc.M110.184010; PMID: 21087933
- Rushworth JV, Griffiths HH, Watt NT, Hooper NM. Prion protein-mediated toxicity of amyloid-β oligomers requires lipid rafts and the transmembrane LRP1. J BiolChem 2013; 288:8935 - 51; http://dx.doi.org/10.1074/jbc.M112.400358; PMID: 23386614
- Um JW, Kaufman AC, Kostylev M, Heiss JK, Stagi M, Takahashi H, Kerrisk ME, Vortmeyer A, Wisniewski T, Koleske AJ, et al. Metabotropic glutamate receptor 5 is a coreceptor for Alzheimer aβ oligomer bound to cellular prion protein. Neuron 2013; 79:887 - 902; http://dx.doi.org/10.1016/j.neuron.2013.06.036; PMID: 24012003