
Abstract
Prions are a novel form of infectivity based on the misfolding of a self-protein (PrPC) into a pathological, infectious isomer (PrPSc). The current uncontrolled spread of chronic wasting disease in cervids, coupled with the demonstrated zoonotic nature of select livestock prion diseases, highlights the urgent need for disease management tools. While there is proof-of-principle evidence for a prion vaccine, these efforts are complicated by the challenges and risks associated with induction of immune responses to a self-protein. Our priority is to develop a PrPSc-specific prion vaccine based on epitopes that are uniquely exposed upon misfolding. These disease specific epitopes (DSEs) have the potential to enable specific targeting of the pathological species through immunotherapy. Here we review outcomes of the translation of a prion DSE into a PrPSc-specific vaccine based on the criteria of immunogenicity, safety and specificity.
Introduction
Transmissible spongiform encephalopathies (TSEs), or prion diseases, are fatal neurodegenerative disorders of both humans and animals. The molecular basis of these proteinopathies resides in the misfolding of a normal cellular protein (PrPC) into a pathological, infectious conformation (PrPSc).Citation1 The “protein-only” hypothesis stipulates that PrPSc serves as a template, or seed, to promote the conversion of PrPC into the infectious conformation in an autocatalytic, self-propagating manner.Citation1-Citation4 While other proteinopathies, such as amyotrophic lateral sclerosis, Alzheimer disease, Parkinson disease and Huntington’s disease share common features of self-propagation, the prion diseases appear at this time to be unique in their ability to transmit across individuals and species in natural settings.Citation1,Citation5-Citation9
Prion diseases have been characterized in a number of species. Among domestic animals this includes scrapie of sheep, bovine spongiform encephalopathy (BSE) of cattle and chronic wasting disease (CWD) of cervids.Citation2-Citation4,Citation10,Citation11 A number of human prion diseases of distinct origins and pathologies have also been characterized. These include iatrogenic, familial and spontaneous forms of the disease as well as prion diseases acquired from consumption of prion infected materials, such as kuru and variant Creutzfeldt-Jakob disease (vCJD).Citation12
Bovine Spongiform Encephalopathy
In the 1980s a mysterious disease outbreak in cattle brought prion diseases to the forefront of public and scientific attention. This disease, termed mad cow disease or BSE, was later identified as a prion disease. Prior to this, prion diseases were typically regarded as either non-threatening to human health (as in the case of scrapie) or diseases of exotic locations and circumstances (as in the case of kuru). BSE served as a grim lesson of how prion diseases can infect domestic herds and in turn threaten human health.
Over the course of the BSE outbreak nearly two hundred thousand animals were confirmed to be prion infected and millions entered into the food chain undetected.Citation13 As a consequence of prion-tainted beef consumption, over two hundred people contracted vCJD, a new form of human prion disease. Fortunately, species barriers, that dictate prion infection transmission efficiency, prevented a far worse outcome.
Chronic Wasting Disease
Chronic Wasting Disease (CWD) is a prion disease of cervids with a host range that includes elk, deer and moose.Citation14,Citation15 Initially described in the 1960s, CWD was later confirmed as a prion disease.Citation16 CWD is considered highly infectious, based on its prevalence (infection rates can reach 30% and 100% in wild and captive cervid populations respectively) and extensive geographic spread (17 US states and two Canadian provinces).Citation17,Citation18
At this time there are no documented cases of CWD transmission to humans. While the report of CJD in three young deer hunters was alarming, the neuropathology of these victims was inconsistent with either vCJD or CWD.Citation19,Citation20 Experimental investigations of CWD’s zoonotic potential are also inconclusive. CWD is transmissible to squirrel monkeys, suggesting the susceptibility of some primates, but it is not transmissible to transgenic mice expressing human PRNP, suggesting that humans may not be at risk.Citation21,Citation22
In assessing the zoonotic potential of CWD it is important to appreciate the restrictions likely imposed by the species barrier and other factors. As mentioned, the relatively low number of people that developed clinical vCJD following the consumption of BSE-contaminated beef highlights the inefficient transmission of prions across species. However, a survey of archived appendixes showed as many as 1 in 2000 residents in the UK may be asymptomatic carriers of abnormal PrP.Citation23 This may be significant for CWD where a combination of low transmissibility to humans, genetic susceptibility, prolonged latency periods, relatively low levels of human consumption of cervid-based foods, and other unknown factors may mask zoonotic potential.
The potential threat of CWD to human health is not limited to direct transmission; indirect transmission through a secondary species, such as cattle, is also a concern.Citation24 The close ecological and phylogenetic relationship of cervids to cattle is consistent with the potential for disease transmission.Citation25 Cattle orally inoculated with CWD-infected brains, or housed with CWD-positive deer, do not contract a TSE but cattle inoculated intracerebrally with CWD do develop a prion disease.Citation18,Citation26 This highlights the potential, given the correct circumstances, for CWD to overcome this species barrier. Prion disease transmission from cervids to cattle could also occur through any number of intermediate species. While it would be speculative to predict the characteristics, including zoonotic potential, of the disease that would occur should CWD ever transmit to cattle it would seem certain that the perception of such a disease would have profound implications for the livestock industry.
While BSE and CWD share a common molecular mechanism, the management of these livestock diseases present very different challenges. Unlike the BSE outbreak, which has been effectively controlled at the level of animal management, it is much more difficult to manage CWD where sources of infection come from, and within, wild animals and/or environmental contamination. To date, the efforts to control CWD, in particular within wild cervid populations, have been largely unsuccessful.Citation27,Citation28
Immunotherapy of Prion Diseases
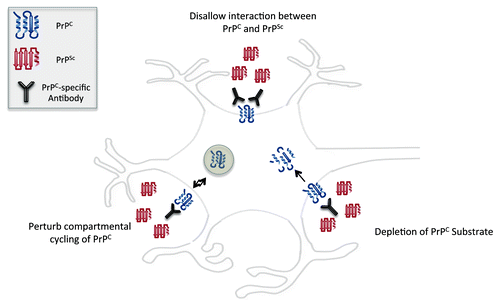
There is clear priority for the development of tools and strategies that enable the control and prevention of prion diseases. There have been numerous attempts to develop a prion vaccine utilizing different antigenic determinants coupled with a variety of formulation and delivery strategies.Citation29-Citation35 While these efforts have not yielded an effective prion vaccine, they have supplied proof-of-principle evidence that antibodies can interfere with disease progression and pathology. This may occur through disruption of PrPC-PrPSc interactions, influencing of the compartmental cycling of PrP or, in the case of antibodies that target PrPC, depletion of the substrate for protein misfolding (). Several comprehensive reviews are available describing approaches and outcomes of prion vaccine development by the general research community.Citation17,Citation29,Citation36 This review will focus on progress in developing conformation-specific peptide vaccines targeting PrPSc.
Figure 1. Effects of PrPC immunotherapy on reducing amplification of PrPSc.
Immunogenicity
Formulation and delivery
Immunological tolerance is a central challenge to the development of a prion vaccine, or any vaccine against a self-molecule. In order to induce immune responses against PrPC, or to specific regions of this protein, other groups have employed a variety of carrier systems and adjuvants.Citation29,Citation37 While these approaches are valid for academic investigations, many of the strategies and reagents employed are inconsistent with a real world human or veterinary vaccine due to their toxicities, costs and/or regulatory restrictions.Citation33,Citation38-Citation43 Our efforts have focused on reagents and approaches (from the perspectives of safety, financial and regulatory constraints) that are compatible with a commercial veterinary vaccine.
Leukotoxin carrier protein
Our efforts to develop an injected prion vaccine have utilized a modified toxoid version of Mannheimia hemolytica leukotoxin (Lkt) as a carrier molecule for prion peptide epitopes. Leukotoxin has proven to be a potent immunogen capable of vectoring a variety of peptide self-epitopes such as GnRH.Citation44-Citation46 In addition, Lkt has considerable advantages in terms of cost, demonstrated safety and efficacy in livestock vaccines, and ease of production and licensing for use in veterinary vaccines.
The Lkt platform has proven to be a safe and efficient platform to induce strong immune responses against various peptide-epitopes of PrP in a variety of species including mice, sheep, cattle, deer and elk. There are, however, limitations to the ability of the Lkt system to overcome self-tolerance. The initial efforts of our group to develop prion vaccines based on select epitopes of PrPC coupled to the Lkt carrier failed to induce epitope-specific immune responses. As such, the Lkt carrier is not a “magic bullet” for delivery of peptide self-epitopes. This highlights the importance of epitope optimization in parallel with efforts to optimize formulation and delivery.
While the Lkt carrier has proven valuable in the context of an injectable vaccine, it is not an ideal carrier for the development of an oral CWD vaccine. While an injected prion vaccine for farmed livestock is a priority, ultimately an oral vaccine will be required to manage the propagation and amplification of prions in wild cervid populations. Our group is investigating alternate carrier proteins that may be more appropriate for an oral prion vaccine.
Disease-specific epitopes
Therapeutic strategies based on the induction of immune responses to self-antigens are complicated by the potential for pathological consequences as a result of autoreactive antibodies. As such there is appropriate concern over the induction of immune responses to PrPC, a widely expressed cell surface protein. These concerns are supported by the observation that PrPC-reactive antibodies induce extensive apoptosis of neurons in the brain and instigate inappropriate cell signal activation and cellular function of suppressor T-cell lymphocytes.Citation47,Citation48
The limited phenotypic consequences associated with genetic deletion of PrPC could be interpreted to predict minimal consequence of autoantibodies to PrPC. However changes to PrPC function as a consequence of antibody binding, particularly from a signaling perspective, may be distinct from the loss-of-function associated with genetic knockouts. Although it is reassuring that there have been no reports of significant pathology associated with the induction of PrPC-reactive immune responses, it is important to appreciate that antibodies to different regions of PrPC may have distinct functional consequences for the target protein. This hypothesis is supported by the recent report of unique outcomes, some of which are pathological, as a result of antibody binding to distinct regions of PrPC.Citation49 As such, the safety of autoreactive antibodies to PrPC is most appropriately evaluated on a case-by-case basis and, ideally, reactivity to PrPC is avoided altogether ().
Figure 2. Effect of conformation-specific immunotherapy on endogenous PrPC and PrPSc.
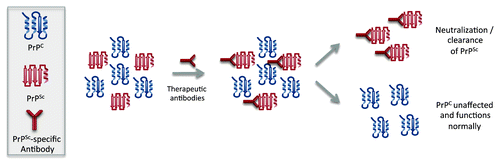
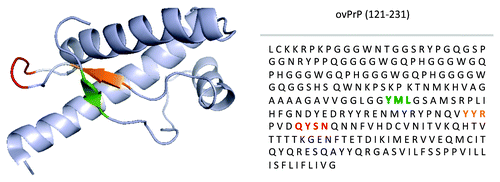
Given the potential pathologies associated with the induction of immune responses to the native conformation of a self-protein, there is considerable appeal to selectively stimulating immune responses to the misfolded species. Conformation-specific immunotherapy is enabled by the unique molecular mechanism of prion diseases whereby the misfolding of PrPC into the pathological conformation results in the surface exposure of protein regions that are normally buried within the native structure. These DSEs represent highly attractive targets for vaccine development. To date, three PrP DSEs have been proposed. Their sequences and positioning with respect to the primary and tertiary structure of PrPC are presented in ().Citation50 While efforts are underway to translate each of these epitopes into vaccines, the scope of this review will be on efforts relating to the YYR epitope.
Figure 3. Mapping of PrPSc DSEs. Location of YML (green), YYR (orange), and RL (red) disease-specific epitopes within tertiary and primary structure of PrP. Adapted from James et al.Citation50
YYR disease-specific epitope
The first DSE of PrP was identified through biophysical investigations of the refolding of PrPC into PrPSc. Specifically, the increased solvent exposure of tyrosine residues as a consequence of misfolding was mapped to a Tyrosine-Tyrosine-Arginine (YYR) epitope of β-strand 2.Citation51 Consistent with DSE priority, antibodies to this epitope immunoprecipitate PrPSc from prion-infected brain homogenate but do not react with PrPC of uninfected brains.Citation52
YML disease-specific epitope
The localization of the YYR DSE to β-strand 2 prompted consideration of whether the opposing β strand, defined by the triplet Tyrosine-Methione-Leucine (YML), would undergo similar surface exposure upon misfolding. Consistent with this hypothesis, antibodies to the YML epitope do not react with healthy cells through flow cytometry but do bind to prion-infected cells.Citation53
Rigid loop (RL) disease-specific epitope
A third PrP DSE was proposed based on the application of the ProMIS algorithm which is designed to identify regions of native structures most likely to unfold into an unstructured conformation.Citation54 This third DSE is situated within a loop between β-strand 2 and α-helix 2. This loop displays unique conformational rigidity in cervid species as compared with prion proteins of other prion susceptible mammals suggesting it may represent a strong candidate for a PrPSc-specific prion vaccine for cervids.Citation55-Citation57
Optimization of disease specific epitopes
The PrP DSEs are highly attractive targets for development of PrPSc-specific vaccines. Unfortunately, translation of these targets into vaccines is complicated by their limited immunogenicity, even when conjugated to potent carrier molecules in the presence of strong adjuvants. Considerable effort is required to identify expansions of the DSEs that capitalize on the theoretical potential of these vaccine targets through the induction of strong immune responses.
Optimization of YYR
The challenges that separate the identification of a DSE from the generation of a functional vaccine became apparent with the YYR DSE. Here, despite a rigorous vaccination regime involving harsh adjuvants, the immune response to the YYR epitope was limited to IgM antibodies.Citation52 The inability to generate strong antibody responses to this epitope through conventional strategies of vaccine formulation and delivery prompted investigation of more sophisticated peptide delivery systems, specifically the Lkt carrier protein. Unfortunately, even presentation of the YYR epitope as a recombinant fusion to Lkt failed to generate an epitope-specific immune response. The inability of this potent peptide delivery system to overcome the limited immunogenicity of the YYR epitope highlights the challenges associated with vaccine development efforts based on DSEs.
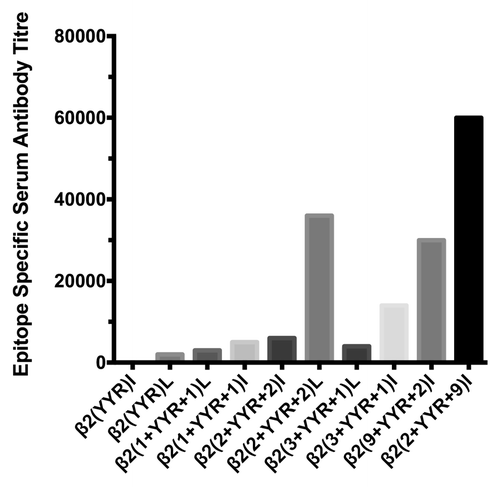
The subsequent effort to improve the immunogenicity of the YYR epitope was an iterative process involving the creation of a panel of vaccines based on expansions of the YYR core to represent the naturally occurring sequence within the PrP protein. These expanded epitopes were presented as recombinant C-terminal fusions of Lkt. This approach was successful as a number of vaccines based on these expansions induced PrPSc-specific antibody responses. The top candidate vaccine, as evaluated by the magnitude of epitope-specific antibody titers, was achieved through the addition of two and nine residues to the N- and C-terminal sides of the YYR core respectively β2(2+YYR+9)I (). This highlighted the potential, but also the effort required, to translate a DSE into a functional vaccine.
Figure 4. Immunogenicity of YYR vaccines in sheep. YYR epitope vaccines were generated as C-terminal fusions to the carrier protein leukotoxin. Epitopes were generated through N and C-terminal expansion of the YYR core, using the PrP protein sequence. In the recombinant protein, epitopes were presented in a repeat motif in either a linear (L) or inverted (I) presentation. Median titers represent serum antibody titers two weeks after a boost immunization.
Specificity
PrPC vs. PrPSc specificity
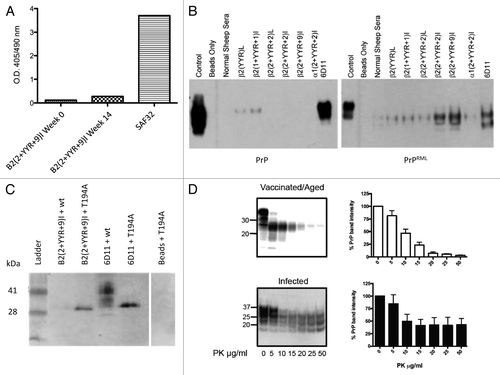
Our efforts to develop a prion vaccine are grounded in the philosophy that a vaccine that induces an immune response that is specific to the pathological form of the protein is advantageous in terms of safety and regulatory potential. As such, it is essential to ensure that the expansions to improve immunogenicity are not at the expense of PrPSc specificity. ELISA experiments utilizing recombinant PrPC confirmed that the β2(2+YYR+9)I vaccine induced responses that are unreactive with PrPC ().Citation24 Immunoprecipitation assays confirmed that these DSE-specific antibodies react with PrPSc but not PrPC ().
Figure 5. Specificity and safety of conformation-specific immunotherapy. (A) Sheep (n = 7) received the β2(2+YYR+9)I vaccine at 6-wk intervals. Pre-immune and immune serum from high titer animals were evaluated for the ability to bind PrPC via ELISA. (B) Sheep were immunized three times at 6-wk intervals with the either of the various YYR-based vaccines noted. Immune sera conjugated to magnetic beads were used in immunoprecipitation experiments with brain homogenates from uninfected (wt) and scrapie-infected mice (RML). (C) Polyclonal β2(2 + YYR + 9)I coupled to beads was used in immunoprecipitation experiments with lysates from HEK293T cells expressing either wt PrPC or the T194A PrPC mutant. (D) PK digests and western blots of combined brain and spleen homogenates from six β2(2 + YYR + 9)I-vaccinated / aged or infected tga20 mice.
Genetic disorders
Our efforts to develop a prion vaccine are motivated by the anticipated administration to cervids for the control of CWD. The conserved molecular mechanism of prion diseases offers the opportunity to apply such a vaccine across a range of species. Further, as the DSE sequences are well conserved across species of potential interest, including humans and cattle, there is the potential to employ the same epitope sequences, or close derivatives, across a range of species.
Within the context of CWD, there is a high probability that cervids will be exposed to the infectious agent. As such, the administration of a prion vaccine to the general cervid population is justified and appropriate. Within human populations, at least under normal circumstances, the risk of exposure to prions is not sufficient to justify the general use of a vaccine. A human prion vaccine may, however, be valuable in instances of increased disease risk, such as individuals working with prions or with suspected exposure to prions, or for individuals with genetic predisposition to prion disease. Familial prion diseases account for 5–15% of human TSE occurrences and include a number of mutations that are associated with increased disease risk.Citation58
The logic to prioritize a prion vaccine to genetically at-risk individuals must be tempered with the appreciation that the same genetic changes that bestow increased disease risk may also influence the specificity and outcomes of PrPSc-specific vaccination. Accordingly, the application of a PrPSc-specific prion vaccine to genetically predisposed subjects requires characterization of the binding proclivities of the induced antibodies within the context of specific PrPC sequence variations.
Reactivity of antibodies induced by the vaccine representing the 2+YYR+9 epitope with wild-type and T194A bovine PrPC were considered to investigate whether mutations associated with increased prion disease susceptibility influence the specificity of PrPSc-specific antibodies. These considerations employed polyclonal antibodies to mimic the physiological outcomes of vaccination, as well as to provide opportunities for binding of the mutant PrPC with a population of antibodies. The T194A mutation is a bovine homolog of the human T183A PrPC mutation that is associated with early onset of familial dementia.Citation59,Citation60 This mutation disrupts glycosylation, promotes structural instability, potentiates misfolding, and accelerates conversion of PrPC to PrPSc.Citation61–Citation63
Nanopore analysis demonstrated that the PrPSc-specific antibody did not react with wild-type PrPC but did bind to T194A under non-denaturing conditions.Citation64 ELISA and immunoprecipitation assays using recombinant T194A protein confirmed the binding to the mutant but not wild-type protein ().
The ability of PrPSc-specific antibodies to bind disease-associated PrP suggests this mutant may have greater propensity to adopt conformations consistent with the disease-associated folding pattern. Similarly, this interaction may reflect greater flexibility of the mutant protein enabling the antibodies to select isoforms presenting the DSE.Citation62 Alternatively, the antibody may serve as a chaperone to induce conformations of the mutant PrPC protein that maximize DSE exposure. Independent of the specific mechanism, the ability of the DSE-specific antibody to bind the mutant PrPC raises concern over the outcomes of PrPSc-specific vaccine administration to individuals with this, or other mutations, that increase propensity for prion disease.
At this time it is critical to determine if the PrPSc-specific antibodies are appropriately recognizing the products of spontaneous misfolding or promoting these misfolding events with the misfolding prone PrP protein. Vaccination experiments in transgenic animals expressing the mutant form of PrPC should address this question as evaluated by whether the vaccinated animals display accelerated or prolonged disease progression, as well as variation in PrPSc deposition profiles. The outcome of these experiments will have implications for the application of DSE–based vaccines in populations predisposed for prion disease.
At this time it would be premature to discount the potential for conformation-specific immunotherapy for individuals with genetic predisposition to prion disease. Further information is required regarding how specific mutations influence the efficacy of the vaccine in protecting from prion pathology, as well as for safety considerations with respect to the antibody’s potential to promote disease through misfolding of PrPC or activation of deleterious cellular responses through PrPC binding. These considerations will require investigation of how specific mutations influence the interactions with antibodies against specific DSEs, as it may be possible to induce PrPSc-specific responses in genetically susceptible individuals by targeting specific DSEs.
Safety
Safety of induced immune responses
Induction of antibodies specific to the misfolded conformation is a rational strategy to circumvent risks associated with the induction of autoreactive antibodies. There are concerns, however, of the potential for these antibodies to initiate template-directed misfolding of PrPC. In a worst-case scenario, a prion vaccine based on targeting the misfolded species could initiate or accelerate a prion disease.
Previous attempts to induce the misfolding of PrPC to PrPSc with antibodies that bind to putative binding and conversion domains increased exposure of select disease-associated motifs but did not generate proteinase K (PK) resistant material.Citation65 These observations suggest that template-directed misfolding has relatively stringent conditions and argues against PrPSc-specific antibodies posing any threat for disease initiation or amplification. Conversely, the ability for PMCA assays to generate misfolded PrP and overcome species barriers implies a margin of flexibility in the template and substrates that can result in directed misfolding.Citation25,Citation66 Collectively, while the probability of antibody-induced misfolding of PrPC is quite low, the potential consequences are important enough to justify investigation.
In vitro template directed misfolding
The ability of DSE-specific antibodies to function as chaperones that promote, or stabilize, the misfolding of PrPC to PrPSc was initially considered within in vitro systems. This included incubation of purified, polyclonal ovine antibodies generated to the YYR DSE with ovine brain homogenates. Following 24 h of shaking at 37 °C there was no evidence of PK resistant material. These results support the conclusion that DSE-specific antibodies are unable to induce template-directed misfolding of PrPC to PK-resistant PrPSc in vitro.
While results of these previous investigations support the inability of PrPSc-specific vaccines to induce misfolding, this must be tempered with the appreciation that these trials were conducted with wild-type PrPC. Given our previous demonstration of the ability of the PrPSc-specific antibodies to bind PrPC mutants associated with familial prion disease, it was appropriate to investigate antibody-induced misfolding of a misfolding-prone PrPC. Incubation of the T194A mutant with the ovine DSE antibodies failed to generate protease resistant species indicating the probability of antibodies inducing misfolding is minimal, even with a substrate with greater structural propensity for misfolding.Citation64
In vivo template directed misfolding
While the inability of the DSE-specific antibodies to promote in vitro misfolding of PrPC is reassuring, it is important to appreciate that such experiments do not replicate the in vivo environment. Therefore, we inquired whether DSE-based vaccines were capable of antibody-induced misfolding of PrPC in sheep. Animals receiving the vaccine were monitored for clinical indications of scrapie as well as generation and deposition of PrPSc material. At six months post vaccination, no clinical indications of scrapie were observed and IHC examination of obex, cerebellum, and rectal lymphoid follicles coupled with ELISA tests for PK-resistant PrPSc in obex and cerebellum were all negative. Similarly, sheep vaccinated with the optimized YYR-based vaccine have not shown any clinical signs of TSE nearly three years after vaccination. Furthermore, elk vaccinated with the same YYR-based vaccine did not show clinical signs of prion disease and post-mortem analysis of obex tissues revealed no detectable PrPSc at one year post-vaccination.
As antibody-induced PrPC misfolding may be a low-frequency event that is difficult to detect in wild-type animals, we investigated the ability of a PrPSc-specific antibody to promote PrPC misfolding in a highly sensitive animal model of prion disease. Homozygous tga20 mice, which carry approximately 60 copies of the PRNP gene and express approximately 6-fold more PrP than wild-type mice, are highly susceptible to the development of prion disease.Citation67 Within this sensitive model, mice received the optimized YYR-based vaccine and were probed for deposition of PK-resistant material more than 8.5 mo post vaccination.Citation68 These mice, and their unvaccinated, age-matched controls, remained healthy with no abnormal behavior within the testing period of 255 d post-immunization (311 d of age) (). There was no evidence for the formation of PK-resistant PrPSc within the brains and spleens of these animals. These results suggest that the vaccination strategy using DSEs is safe in the context of an aggressive in vivo model of TSE that heavily favors disease, and also supports the conclusion that these PrPSc- and DSE-specific antibodies are unable to induce template-directed misfolding.
Vaccine efficacy
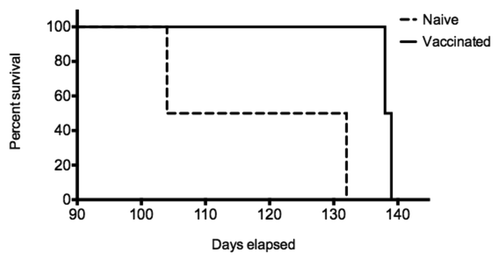
The protective efficacy of the PrPSc-specific antibodies was evaluated in a sheep oral challenge model. Pregnant ewes were immunized with either the Lkt-β2(2+YYR+2)I vaccine or the Lkt carrier (n = 2/group). Immediately after birth lambs were challenged via gastric feeding with scrapie material. Newborn lambs then suckled, transferring maternal PrPSc-specific antibodies from vaccinated ewes to lambs. Lambs born, and suckling, of Lkt-β2(2+YYR+2)I vaccinated mothers (n = 3) had a significantly delayed onset of disease in comparison to lambs nursed by Lkt vaccinated mothers (n = 4) (). This confirms the ability of the antibodies to neutralize the challenge dose in vivo. While a small study, the observed protection is statistically significant (P < 0.019) and offers clear support for the concept of PrPSc-speicific immunotherapy. Furthermore, this protection was achieved in an aggressive disease model, with an early generation vaccine that induces responses that are two orders of magnitude lower than our currently most immunogenic vaccine. Challenge trials with these optimized vaccines are underway.
Figure 6. Active immunization and oral challenge of sheep. Pregnant ewes (n = 2/group) received two immunizations of either Lkt or Lkt-β2(2+YYR+9)I vaccine at six and three weeks prior to birthing. Lambs born from Lkt (n = 4) or Lkt-β2(2 + YYR + 9)I (n = 3) vaccinated ewes were challenged with 1 g of scrapie brain homogenate via gastric feeding immediately following birth. Animals were continuously monitored for the onset of scrapie symptoms by 24 h video surveillance.
Conclusions
Our group has focused on the development of a vaccine to control prion diseases within animals, focusing on cervids. While an immunotherapeutic strategy is not a novel concept to manage prion diseases, our approach seeks to immunologically discriminate between normal and infectious PrP conformers, potentially mitigating safety issues that arise when targeting a self-protein.
Following the generation of candidate vaccines, it is necessary to validate construct immunogenicity, as well as specificity of induced antibodies for the misfolded species of PrP. While the absence of antibody reactivity with the properly folded species offers reassurance of vaccine safety, it is still essential to confirm that the induced antibodies do not serve as chaperones to promote misfolding events or other pathologies in vivo. The vaccine generated and evaluated within this project has potential for an excellent safety profile due to the absence of pathological clinical symptoms in vaccinated animals, including highly sensitive transgenic models of prion disease, the minimal risk of autoreactivity induction, and the inability to detect the generation of PrPSc.
The question of specificity is complicated in instances where genetic mutations confer increased susceptibility to disease, as these mutated species may have greater propensity for natural and induced exposure of misfolding-specific epitopes. These specific scenarios will require consideration for potential future applications of a PrPSc-specific vaccine for the prevention and/or treatment of familial forms of prion disease but are not anticipated as a critical consideration within the context of a veterinary vaccine.
The ultimate test of these vaccines, and the concept of disease-specific immunotherapy for prion diseases, will be the ability to exert a therapeutic effect in animal challenge models. There are a number of potential mechanisms and outcomes through which these vaccines may emerge as an important tool for the control of prion diseases. Most notably the ability to protect animals from infection in relevant challenge models (in terms of the dose and route of challenge material) would be a landmark success. Success could also take the form of providing a tool to protect animal populations, rather than individual animals, through the reduced generation and shedding of infectious materials. Preliminary experiments have demonstrated the ability of a YYR-based, PrPSc-specific vaccine to delay the onset of symptoms in a large animal prion infection model. Further experiments to determine the ability of these vaccines to influence disease initiation, progression and transmission have been initiated to address these critical questions.
| Abbreviations: | ||
| DSE | = | disease-specific epitope |
| TSE | = | transmissible spongiform encephalopathy |
| BSE | = | bovine spongiform encephalopathy |
| CWD | = | chronic wasting disease |
| (v)CJD | = | (variant) Creutzfeldt-Jakob disease |
| Lkt | = | leukotoxin |
| YYR | = | tyrosine-tyrosine-arginine |
Disclosure of Potential Conflicts of Interest
Neil Cashman is the Founder and CSO of Amorfix Life Sciences.
Acknowledgments
This work was funded by PrioNet Canada and Alberta Prion Research Institute.
Related Research Data
References
- Prusiner SB. Molecular biology of prion diseases. Science 1991; 252:1515 - 22; http://dx.doi.org/10.1126/science.1675487; PMID: 1675487
- Griffith JS. Self-replication and scrapie. Nature 1967; 215:1043 - 4; http://dx.doi.org/10.1038/2151043a0; PMID: 4964084
- Prusiner SB. Novel proteinaceous infectious particles cause scrapie. Science 1982; 216:136 - 44; http://dx.doi.org/10.1126/science.6801762; PMID: 6801762
- Aguzzi A, Sigurdson C, Heikenwaelder M. Molecular mechanisms of prion pathogenesis. Annu Rev Pathol 2008; 3:11 - 40; http://dx.doi.org/10.1146/annurev.pathmechdis.3.121806.154326; PMID: 18233951
- Hunter N, Houston F, Foster J, Goldmann W, Drummond D, Parnham D, Kennedy I, Green A, Stewart P, Chong A. Susceptibility of young sheep to oral infection with bovine spongiform encephalopathy decreases significantly after weaning. J Virol 2012; 86:11856 - 62; http://dx.doi.org/10.1128/JVI.01573-12; PMID: 22915816
- Kimberlin RH, Wilesmith JW. Bovine spongiform encephalopathy. Epidemiology, low dose exposure and risks. Ann N Y Acad Sci 1994; 724:210 - 20; http://dx.doi.org/10.1111/j.1749-6632.1994.tb38911.x; PMID: 8030941
- Mathiason CK, Powers JG, Dahmes SJ, Osborn DA, Miller KV, Warren RJ, Mason GL, Hays SA, Hayes-Klug J, Seelig DM, et al. Infectious prions in the saliva and blood of deer with chronic wasting disease. Science 2006; 314:133 - 6; http://dx.doi.org/10.1126/science.1132661; PMID: 17023660
- Jeffrey M, Ryder S, Martin S, Hawkins SA, Terry L, Berthelin-Baker C, Bellworthy SJ. Oral inoculation of sheep with the agent of bovine spongiform encephalopathy (BSE). 1. Onset and distribution of disease-specific PrP accumulation in brain and viscera. J Comp Pathol 2001; 124:280 - 9; http://dx.doi.org/10.1053/jcpa.2001.0465; PMID: 11437504
- Race R, Jenny A, Sutton D. Scrapie infectivity and proteinase K-resistant prion protein in sheep placenta, brain, spleen, and lymph node: implications for transmission and antemortem diagnosis. J Infect Dis 1998; 178:949 - 53; http://dx.doi.org/10.1086/515669; PMID: 9806020
- Silveira JR, Caughey B, Baron GS. Prion Protein and the Molecular Features of Transmissible Spongiform Encephalopathy Agents. In: … Cow Disease and Related Spongiform…. Berlin, Heidelberg: Springer Berlin Heidelberg; 2004. pages 1–50.
- van Raamsdonk LWD. Holst von C, Baeten V, Berben G, Boix A, de Jong J. New developments in the detection and identification of processed animal proteins in feeds. Anim Feed Sci Technol 2007; 133:63 - 83; http://dx.doi.org/10.1016/j.anifeedsci.2006.08.004
- Collinge J. Prion diseases of humans and animals: their causes and molecular basis. Annu Rev Neurosci 2001; 24:519 - 50; http://dx.doi.org/10.1146/annurev.neuro.24.1.519; PMID: 11283320
- Nathanson N, Wilesmith J, Griot C. Bovine spongiform encephalopathy (BSE): causes and consequences of a common source epidemic. Am J Epidemiol 1997; 145:959 - 69; http://dx.doi.org/10.1093/oxfordjournals.aje.a009064; PMID: 9169904
- Williams ES. Chronic wasting disease. Vet Pathol 2005; 42:530 - 49; http://dx.doi.org/10.1354/vp.42-5-530; PMID: 16145200
- Sigurdson CJ. A prion disease of cervids: chronic wasting disease. Vet Res 2008; 39:41; http://dx.doi.org/10.1051/vetres:2008018; PMID: 18381058
- Aguzzi A. Staining, straining and restraining prions. Nat Neurosci 2008; 11:1239 - 40; http://dx.doi.org/10.1038/nn1108-1239; PMID: 18956007
- Li L, Napper S, Cashman NR. Immunotherapy for prion diseases: opportunities and obstacles. Immunotherapy 2010; 2:269 - 82; http://dx.doi.org/10.2217/imt.10.3; PMID: 20635933
- Williams ES, Miller MW, Kreeger TJ, Kahn RH, Thorne ET. Chronic wasting disease of deer and elk: a review with recommendations for management. J Wildl Manage 2002; 66:551 - 63; http://dx.doi.org/10.2307/3803123
- Belay ED, Gambetti P, Schonberger LB, Parchi P, Lyon DR, Capellari S, McQuiston JH, Bradley K, Dowdle G, Crutcher JM, et al. Creutzfeldt-Jakob disease in unusually young patients who consumed venison. Arch Neurol 2001; 58:1673 - 8; http://dx.doi.org/10.1001/archneur.58.10.1673; PMID: 11594928
- Belay ED, Maddox RA, Williams ES, Miller MW, Gambetti P, Schonberger LB. Chronic wasting disease and potential transmission to humans. Emerg Infect Dis 2004; 10:977 - 84; http://dx.doi.org/10.3201/eid1006.031082; PMID: 15207045
- Kong Q, Huang S, Zou W, Vanegas D, Wang M, Wu D, Yuan J, Zheng M, Bai H, Deng H, et al. Chronic wasting disease of elk: transmissibility to humans examined by transgenic mouse models. J Neurosci 2005; 25:7944 - 9; http://dx.doi.org/10.1523/JNEUROSCI.2467-05.2005; PMID: 16135751
- Race B, Meade-White KD, Miller MW, Barbian KD, Rubenstein R, LaFauci G, Cervenakova L, Favara C, Gardner D, Long D, et al. Susceptibilities of nonhuman primates to chronic wasting disease. Emerg Infect Dis 2009; 15:1366 - 76; http://dx.doi.org/10.3201/eid1509.090253; PMID: 19788803
- Gill ON, Spencer Y, Richard-Loendt A, Kelly C, Dabaghian R, Boyes L, Linehan J, Simmons M, Webb P, Bellerby P, et al. Prevalent abnormal prion protein in human appendixes after bovine spongiform encephalopathy epizootic: large scale survey. BMJ 2013; 347:f5675; http://dx.doi.org/10.1136/bmj.f5675; PMID: 24129059
- Hedlin P, Taschuk R, Potter A, Griebel P, Napper S. Detection and control of prion diseases in food animals. ISRN Vet Sci 2012; 2012:254739; http://dx.doi.org/10.5402/2012/254739; PMID: 23738120
- Castilla J, Gonzalez-Romero D, Saá P, Morales R, De Castro J, Soto C. Crossing the species barrier by PrP(Sc) replication in vitro generates unique infectious prions. Cell 2008; 134:757 - 68; http://dx.doi.org/10.1016/j.cell.2008.07.030; PMID: 18775309
- Hamir AN, Kehrli ME Jr., Kunkle RA, Greenlee JJ, Nicholson EM, Richt JA, Miller JM, Cutlip RC. Experimental interspecies transmission studies of the transmissible spongiform encephalopathies to cattle: comparison to bovine spongiform encephalopathy in cattle. J Vet Diagn Invest 2011; 23:407 - 20; http://dx.doi.org/10.1177/1040638711403404; PMID: 21908269
- Almberg ES, Cross PC, Johnson CJ, Heisey DM, Richards BJ. Modeling routes of chronic wasting disease transmission: environmental prion persistence promotes deer population decline and extinction. PLoS One 2011; 6:e19896; http://dx.doi.org/10.1371/journal.pone.0019896; PMID: 21603638
- Wasserberg G, Osnas EE, Rolley RE, Samuel MD. Host culling as an adaptive management tool for chronic wasting disease in white-tailed deer: a modelling study. J Appl Ecol 2009; 46:457 - 66; http://dx.doi.org/10.1111/j.1365-2664.2008.01576.x; PMID: 19536340
- Roettger Y, Du Y, Bacher M, Zerr I, Dodel R, Bach J-P. Immunotherapy in prion disease. Nat Rev Neurol 2013; 9:98 - 105; http://dx.doi.org/10.1038/nrneurol.2012.258; PMID: 23247613
- Enari M, Flechsig E, Weissmann C. Scrapie prion protein accumulation by scrapie-infected neuroblastoma cells abrogated by exposure to a prion protein antibody. Proc Natl Acad Sci U S A 2001; 98:9295 - 9; http://dx.doi.org/10.1073/pnas.151242598; PMID: 11470893
- Perrier V, Solassol J, Crozet C, Frobert Y, Mourton-Gilles C, Grassi J, Lehmann S. Anti-PrP antibodies block PrPSc replication in prion-infected cell cultures by accelerating PrPC degradation. J Neurochem 2004; 89:454 - 63; http://dx.doi.org/10.1111/j.1471-4159.2004.02356.x; PMID: 15056288
- White AR, Hawke SH. Immunotherapy as a therapeutic treatment for neurodegenerative disorders. J Neurochem 2003; 87:801 - 8; http://dx.doi.org/10.1046/j.1471-4159.2003.02064.x; PMID: 14622111
- Sigurdsson EM, Brown DR, Daniels M, Kascsak RJ, Kascsak R, Carp R, Meeker HC, Frangione B, Wisniewski T. Immunization delays the onset of prion disease in mice. Am J Pathol 2002; 161:13 - 7; http://dx.doi.org/10.1016/S0002-9440(10)64151-X; PMID: 12107084
- Sigurdsson EM, Sy M-S, Li R, Scholtzova H, Kascsak RJ, Kascsak R, Carp R, Meeker HC, Frangione B, Wisniewski T. Anti-prion antibodies for prophylaxis following prion exposure in mice. Neurosci Lett 2003; 336:185 - 7; http://dx.doi.org/10.1016/S0304-3940(02)01192-8; PMID: 12505623
- Peretz D, Williamson RA, Kaneko K, Vergara J, Leclerc E, Schmitt-Ulms G, Mehlhorn IR, Legname G, Wormald MR, Rudd PM, et al. Antibodies inhibit prion propagation and clear cell cultures of prion infectivity. Nature 2001; 412:739 - 43; http://dx.doi.org/10.1038/35089090; PMID: 11507642
- Müller-Schiffmann A, Korth C. Vaccine approaches to prevent and treat prion infection : progress and challenges. BioDrugs 2008; 22:45 - 52; http://dx.doi.org/10.2165/00063030-200822010-00005; PMID: 18215090
- Trevitt CR, Collinge J. A systematic review of prion therapeutics in experimental models. Brain 2006; 129:2241 - 65; http://dx.doi.org/10.1093/brain/awl150; PMID: 16816391
- Hanan E, Goren O, Eshkenazy M, Solomon B. Immunomodulation of the human prion peptide 106-126 aggregation. Biochem Biophys Res Commun 2001; 280:115 - 20; http://dx.doi.org/10.1006/bbrc.2000.4097; PMID: 11162487
- Koller MF, Grau T, Christen P. Induction of antibodies against murine full-length prion protein in wild-type mice. J Neuroimmunol 2002; 132:113 - 6; http://dx.doi.org/10.1016/S0165-5728(02)00316-8; PMID: 12417440
- Rosset MB, Ballerini C, Grégoire S, Metharom P, Carnaud C, Aucouturier P. Breaking immune tolerance to the prion protein using prion protein peptides plus oligodeoxynucleotide-CpG in mice. J Immunol 2004; 172:5168 - 74; PMID: 15100253
- Polymenidou M, Heppner FL, Pellicioli EC, Urich E, Miele G, Braun N, Wopfner F, Schätzl HM, Becher B, Aguzzi A. Humoral immune response to native eukaryotic prion protein correlates with anti-prion protection. Proc Natl Acad Sci U S A 2004; 101:Suppl 2 14670 - 6; http://dx.doi.org/10.1073/pnas.0404772101; PMID: 15292505
- Schwarz A, Krätke O, Burwinkel M, Riemer C, Schultz J, Henklein P, Bamme T, Baier M. Immunisation with a synthetic prion protein-derived peptide prolongs survival times of mice orally exposed to the scrapie agent. Neurosci Lett 2003; 350:187 - 9; http://dx.doi.org/10.1016/S0304-3940(03)00907-8; PMID: 14550926
- Gilch S, Schätzl HM. Promising developments bringing prion diseases closer to therapy and prophylaxis. Trends Mol Med 2003; 9:367 - 9; http://dx.doi.org/10.1016/S1471-4914(03)00144-8; PMID: 13129701
- Gerdts V, Mutwiri G, Richards J, van Drunen Littel-van den Hurk S, Potter AA. Carrier molecules for use in veterinary vaccines. Vaccine 2013; 31:596 - 602; http://dx.doi.org/10.1016/j.vaccine.2012.11.067; PMID: 23219438
- Manns JG, Barker C, Attah-Poku SK. The design, production, purification, and testing of a chimeric antigen protein to be used as an immunosterilant in domestic animals. Can J Chem 1997; 75:829 - 33; http://dx.doi.org/10.1139/v97-100
- Hedlin PD, Cashman NR, Li L, Gupta J, Babiuk LA, Potter AA, Griebel P, Napper S. Design and delivery of a cryptic PrP(C) epitope for induction of PrP(Sc)-specific antibody responses. Vaccine 2010; 28:981 - 8; http://dx.doi.org/10.1016/j.vaccine.2009.10.134; PMID: 19925901
- Solforosi L, Criado JR, McGavern DB, Wirz S, Sánchez-Alavez M, Sugama S, DeGiorgio LA, Volpe BT, Wiseman E, Abalos G, et al. Cross-linking cellular prion protein triggers neuronal apoptosis in vivo. Science 2004; 303:1514 - 6; http://dx.doi.org/10.1126/science.1094273; PMID: 14752167
- Mouillet-Richard S, Ermonval M, Chebassier C, Laplanche JL, Lehmann S, Launay JM, Kellermann O. Signal transduction through prion protein. Science 2000; 289:1925 - 8; http://dx.doi.org/10.1126/science.289.5486.1925; PMID: 10988071
- Sonati T, Reimann RR, Falsig J, Baral PK, O’Connor T, Hornemann S, Yaganoglu S, Li B, Herrmann US, Wieland B, et al. The toxicity of antiprion antibodies is mediated by the flexible tail of the prion protein. Nature 2013; 501:102 - 6; http://dx.doi.org/10.1038/nature12402; PMID: 23903654
- James TL, Liu H, Ulyanov NB, Farr-Jones S, Zhang H, Donne DG, Kaneko K, Groth D, Mehlhorn I, Prusiner SB, et al. Solution structure of a 142-residue recombinant prion protein corresponding to the infectious fragment of the scrapie isoform. Proc Natl Acad Sci U S A 1997; 94:10086 - 91; http://dx.doi.org/10.1073/pnas.94.19.10086; PMID: 9294167
- Zou W-Q, Cashman NR. Acidic pH and detergents enhance in vitro conversion of human brain PrPC to a PrPSc-like form. J Biol Chem 2002; 277:43942 - 7; http://dx.doi.org/10.1074/jbc.M203611200; PMID: 12161431
- Paramithiotis E, Pinard M, Lawton T, LaBoissiere S, Leathers VL, Zou W-Q, Estey LA, Lamontagne J, Lehto MT, Kondejewski LH, et al. A prion protein epitope selective for the pathologically misfolded conformation. Nat Med 2003; 9:893 - 9; http://dx.doi.org/10.1038/nm883; PMID: 12778138
- Cashman NR. Antibodies And Epitopes Specific To Misfolded Prion Protein. 2010; United States patent US 20120107321.
- Guest W, Cashman N, Plotkin S. Structure-Based Prediction of Unstable Regions in Proteins: Applications to Protein Misfolding Diseases. Bull Am Phys Soc 2009; 54
- Sigurdson CJ, Aguzzi A. Chronic wasting disease. Biochim Biophys Acta 2007; 1772:610 - 8; http://dx.doi.org/10.1016/j.bbadis.2006.10.010; PMID: 17223321
- Gossert AD, Bonjour S, Lysek DA, Fiorito F, Wüthrich K. Prion protein NMR structures of elk and of mouse/elk hybrids. Proc Natl Acad Sci U S A 2005; 102:646 - 50; http://dx.doi.org/10.1073/pnas.0409008102; PMID: 15647363
- Soto C. Constraining the loop, releasing prion infectivity. Proc Natl Acad Sci U S A 2009; 106:10 - 1; http://dx.doi.org/10.1073/pnas.0811625106; PMID: 19118191
- Kovacs GG, Zerbi P, Voigtländer T, Strohschneider M, Trabattoni G, Hainfellner JA, Budka H. The prion protein in human neurodegenerative disorders. Neurosci Lett 2002; 329:269 - 72; http://dx.doi.org/10.1016/S0304-3940(02)00668-7; PMID: 12183028
- Nitrini R, Rosemberg S, Passos-Bueno MR, da Silva LS, Iughetti P, Papadopoulos M, Carrilho PM, Caramelli P, Albrecht S, Zatz M, et al. Familial spongiform encephalopathy associated with a novel prion protein gene mutation. Ann Neurol 1997; 42:138 - 46; http://dx.doi.org/10.1002/ana.410420203; PMID: 9266722
- Kiachopoulos S, Bracher A, Winklhofer KF, Tatzelt J. Pathogenic mutations located in the hydrophobic core of the prion protein interfere with folding and attachment of the glycosylphosphatidylinositol anchor. J Biol Chem 2005; 280:9320 - 9; http://dx.doi.org/10.1074/jbc.M412525200; PMID: 15591591
- Chebaro Y, Derreumaux P. The conversion of helix H2 to beta-sheet is accelerated in the monomer and dimer of the prion protein upon T183A mutation. J Phys Chem B 2009; 113:6942 - 8; http://dx.doi.org/10.1021/jp900334s; PMID: 19371053
- van der Kamp MW, Daggett V. Pathogenic mutations in the hydrophobic core of the human prion protein can promote structural instability and misfolding. J Mol Biol 2010; 404:732 - 48; http://dx.doi.org/10.1016/j.jmb.2010.09.060; PMID: 20932979
- Rogers M, Taraboulos A, Scott M, Groth D, Prusiner SB. Intracellular accumulation of the cellular prion protein after mutagenesis of its Asn-linked glycosylation sites. Glycobiology 1990; 1:101 - 9; http://dx.doi.org/10.1093/glycob/1.1.101; PMID: 1983782
- Madampage CA, Määttänen P, Marciniuk K, Brownlie R, Andrievskaia O, Potter A, Cashman NR, Lee JS, Napper S. Binding of bovine T194A PrP(C) by PrP(Sc)-specific antibodies: potential implications for immunotherapy of familial prion diseases. Prion 2013; 7:301 - 11; http://dx.doi.org/10.4161/pri.25148; PMID: 23787697
- Li L, Guest W, Huang A, Plotkin SS, Cashman NR. Immunological mimicry of PrPC-PrPSc interactions: antibody-induced PrP misfolding. Protein Eng Des Sel 2009; 22:523 - 9; http://dx.doi.org/10.1093/protein/gzp038; PMID: 19602568
- Chianini F, Fernández-Borges N, Vidal E, Gibbard L, Pintado B, de Castro J, Priola SA, Hamilton S, Eaton SL, Finlayson J, et al. Rabbits are not resistant to prion infection. Proc Natl Acad Sci U S A 2012; 109:5080 - 5; http://dx.doi.org/10.1073/pnas.1120076109; PMID: 22416127
- Fischer M, Rülicke T, Raeber A, Sailer A, Moser M, Oesch B, Brandner S, Aguzzi A, Weissmann C. Prion protein (PrP) with amino-proximal deletions restoring susceptibility of PrP knockout mice to scrapie. EMBO J 1996; 15:1255 - 64; PMID: 8635458
- Määttänen P, Taschuk R, Ross L, Marciniuk K, Bertram L, Potter A, Cashman NR, Napper S. PrP(Sc)-specific antibodies do not induce prion disease or misfolding of PrP(C) in highly susceptible Tga20 mice. Prion 2013; 7:434 - 9; http://dx.doi.org/10.4161/pri.26639; PMID: 24105298