
Abstract
Following the discovery of a causal link between bovine spongiform encephalopathy (BSE) in cattle and variant Creutzfeldt–Jakob disease (vCJD) in humans, several experimental approaches have been used to try to assess the potential risk of transmission of other animal transmissible spongiform encephalopathies (TSEs) to humans. Experimental challenge of non-human primates, humanised transgenic mice and cell-free conversion systems have all been used as models to explore the susceptibility of humans to animal TSEs. In this review we compare and contrast in vivo and in vitro evidence of the zoonotic risk to humans from sheep, cattle and deer prions, focusing primarily on chronic wasting disease and our own recent studies using protein misfolding cyclic amplification.
Introduction
Transmissible spongiform encephalopathies (TSEs) or prion diseases are fatal transmissible neurodegenerative conditions affecting humans and other mammals. TSEs are thought to result from a change in the conformation of the normal prion protein (termed PrPC) into an abnormal form of the protein termed PrPSc.Citation1 They have been described in a range of farmed, captive and free-ranging animal species, including bovine spongiform encephalopathy (BSE) in cattle, scrapie in sheep and chronic wasting disease (CWD) in deer and elk. In humans, TSEs occur as sporadic, genetic and acquired diseases, the most common of which is sporadic Creutzfeldt-Jakob disease (sCJD). TSE pathology is characterized by neurological degeneration and death, gliosis and the deposition of PrPSc in the brain. TSEs are (as the name indicates) transmissible, and the causal infectious agents are termed prions.Citation2
Over the past 30 years, substantial efforts have been made to establish the biology of prions. In 1967 the first theoretical model of a self-replication scrapie agent was proposed by Griffith and coworkers.Citation3 In the same year, Pattison and coworkers considered the nature of sheep prions, suggesting a proteinaceous nature.Citation4 At the beginning of the 1980s Stanley Prusiner coined the term “prion”, distinguishing the infectious agent causing TSE from viruses and viroids, and proposing a protein only mechanism of agent replication, which is now known as the “prion hypothesis.”Citation2
The increasing evidence in favor of the prion hypothesis has involved epidemiological, clinical and pathological research on TSEs in human and animals. Increasingly sophisticated experimental models, including non-human primates, wild-type and humanized transgenic mice and cell-free molecular conversion systems, have each contributed to a better understanding of the nature of prions.
Transgenic Mouse Models
Five years after the initial development of transgenic animals, the cellular gene which encodes the prion protein in mammals (PRNP in humans, Prnp in other animals) was identified.Citation5 Five years later, a transgenic murine model with a single amino acid substitution (P101L) in the murine prion protein gene was created. This transgenic mouse model showed a neurodegenerative process, similar to that in humans affected by a corresponding P102L mutation in the PRNP gene.Citation6 This suggested that a human prion disease can be genetically modeled in animals and introduced the use of transgenic animals into the TSE research, although subsequent studies using this model have shown that the relationship between PrPSc formation, neurodegeneration and prion infectivity is not a straightforward one.Citation7 In order to evaluate the association between Prnp gene expression and prion transmission, Bueler and coworkers intracerebrally challenged Prnp knockout mice with a high dose of scrapie prions. The animals failed to develop any clinical signs of a TSE, and showed no TSE pathology in the brain. The authors concluded that development of clinical signs and pathology is strictly dependent on the presence of PrPC, and that incubation time and disease progression are inversely related to the levels of PrPC.Citation8
Transgenic animals have not only been relevant to understanding the biology of prions, they have also contributed to our understanding of their transmission between species. In this regard, the concepts of strain and species barrier acquire critical importance. Briefly, a prion strain is defined as an infectious isolate that causes a distinct prion disease phenotype (incubation time, lesion profile, etc.), which is maintained upon serial transmission when it is inoculated to another host.Citation9 A species barrier can be defined as the difficulty in transmitting a prion disease from one species to another in a primary transmission. In practice this can be recognized by an adaptation of the agent to the second species, producing a shorter incubation periods and uniform transmission properties in a secondary transmission in the second species. The ability of a prion strain to be transmitted to a new host is affected by the differences in primary sequence of the prion protein between donor and host species.Citation10 Additionally, it is affected by the conformational properties associated with the PrPSc.Citation11
In Vitro Conversion Systems
Caughey and coworkers developed an in vitro system in which recombinant hamster PrPC, incubated with the misfolded form of the prion protein and a chaotropic agent, generated “new” protease-resistance material derived from the original PrPC.Citation12 Although this represented a major development in prion biology, a large excess of input material (PrPSc > 50 fold) was required to perform the conversion of the normal prion protein, placing constraints on its utility.
Subsequently, and based on the nucleation and polymerization model, Soto and colleagues described an in vitro procedure that mimics the in vivo PrPSc conversion process. This approach allows amplification of minute quantities of PrPSc in a sample, and is more efficient than the previous cell-free conversion assay reported by Caughey and coworkers. The technique, called protein misfolding cyclic amplification (PMCA), consists of cycles of sonication and incubation periods of a mixture of a normal brain homogenate (which is rich in PrPC) and a source of PrPSc, in the presence of detergents ().Citation13
Figure 1. Schematic representation of protein misfolding cyclic amplification (PMCA) showing the presumed mechanism of molecular conversion. A large excess of PrPC is incubated with minimal quantities of PrPSc followed by cycles of incubation and sonication. PrPSc aggregates are shown to grow, converting and incorporating PrPC molecules. With each cycle of sonication and incubation (represented by arrows) PrPSc aggregates are broken, creating further seeds. After several cycles of sonication and incubation most of the PrPC will have been converted into the PrPSc.
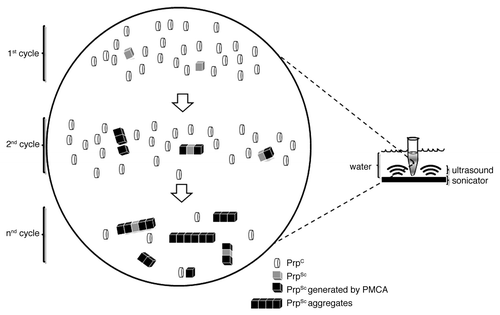
PMCA has proved to be a rapid, versatile and inexpensive method to investigate fundamental molecular aspects of prion protein conversion. For example, a species barrier is known to exist for sheep scrapie strains adapted to mice and to hamsters. Castilla and coworkers reported the use of murine scrapie strains and hamster PrPC to mimic the in vivo adaptation process in vitro, generating new prions by serial rounds of PMCA. Similar results were obtained using hamster seeds and murine substrate.Citation14 This provided the proof of concept that PMCA can recapitulate a biological process in days what would normally take months or even years in vivo.
vCJD and BSE: The First Example of a Zoonotic Prion Disease
In 1985 the emergence of a new TSE affecting cattle was reported in the United Kingdom. The disease, bovine spongiform encephalopathy or BSE, eventually affected more than 180 000 animals in the United Kingdom with more than 70 000 confirmed clinical cases between 1992 and 1993.Citation15 The affected animals suffered a slow progressive neurological disorder, changes in sensation, posture and movement, weight loss and reduced milk yield. Affected cattle were pathologically characterized by brain stem gray matter vacuolation.Citation16
At this time it was unclear whether BSE would prove a threat to human health. BSE is now known to be the pathogen that causes variant CJD (vCJD) in humans, whereas scrapie in sheep is thought to pose little or no risk to human health. In retrospect, the connection between BSE and vCJD seems obvious, but at the time it required careful surveillance and research for the connection to be established.
In 1996 Will and colleagues reported the identification of 10 unusual cases of CJD in young individuals in the UK. Active surveillance identified no obvious increase in the incidence of CJD in young patients in other European countries. Taking into account the young age at onset of the reported cases, a distinctive neropathological profile, the presence of “florid” PrP plaques and the apparent absence of cases with a similar profile in Europe, the authors suggested that the appearance of vCJD in the UK was linked to the earlier BSE epidemic.Citation17
All definite cases of vCJD that have undergone PRNP analysis are methionine homozygotes at codon 129.Citation18 PRNP polymorphisms appear to influence clinical and pathological phenotypes in the human prion diseases. The most important polymorphism resides at codon 129, which results in three polymorphic combinations: the homozygous methionine/methionine and valine/valine, and the heterozygous methionine/valine.
In order to test the link between BSE and vCJD, non-human primates were intracerebral inoculated with BSE brain homogenate. All three BSE-inoculated Cynomolgus macaques developed abnormal behavior similar to that observed in the vCJD cases and had a neuropathology including PrPSc deposition reminiscent of that seen in vCJD patients. This supported the hypothesis that BSE is the agent responsible for vCJD.Citation19 Transmission studies in inbred mouse strains reinforced this view, and strongly suggested that vCJD is caused by the same strain of agent that causes BSE, feline spongiform encephalopathy (FSE) and TSEs in exotic ruminants.Citation20,Citation21 More compiling evidence of a link between BSE and vCJD was presented by Scott and coworkers who inoculated bovinized transgenic mice with BSE, scrapie, and vCJD.Citation22All three inocula propagated efficiently. BSE and vCJD were indistinguishable in terms of incubation time and neuropathology, but were distinct from scrapie, reinforcing the conclusion that vCJD is caused by the same strain of agent as that causing BSE.
In order to model the transmissibility of vCJD, sCJD and BSE to humans, transgenic animals that expressed human PRNP valine (V)Citation23 or methionine (M)Citation24 at codon 129 were inoculated. The transgenic mice presented evidence of transmission for each inoculum. However, the molecular signature of the vCJD-PrPSc generated by the 129–V infected animals did not share the same electrophoretic mobility of the original inoculum.Citation23 In contrast, the animals carrying the polymorphism 129-M (inoculated with vCJD) conserved the molecular pattern of the original inoculum.Citation24 To further evaluate the transmissibility of BSE and vCJD and to assess the effect of codon 129 polymorphism on human susceptibility, Manson and coworkers produced “knock in” humanized transgenic mice carrying the three PRNP codon 129 genotypes: methionine and valine homozygous and methionine/valine heterozygous. After inoculation with BSE and vCJD prions, different pathological characteristics and transmission efficiency was detected among the three genotypes. In terms of vCJD propagation, the MM genotype at codon 129 was more susceptible than MV, which in turn was more susceptible than VV.Citation25 The authors cited a previous publication in which a cell-free assay showed that human purified PrPC with M at position 129 was converted to PrPSc more readily by vCJD and BSE than the 129-V equivalent.Citation26
In order to test whether inter-species transmission barriers could be modeled using PMCA, Jones and coworkers tested the ability of BSE and scrapie to convert human prion protein of the three major human prion protein polymorphic variants (PRNP codon 129 MM, MV and VV) expressed in humanised transgenic mouse brain. The results showed that cattle BSE effected efficient conversion of human PrP with a human PRNP genotypic preference similar to that of human vCJD (MM > MV > VV) and that scrapie failed to convert the human substrate.Citation27 These results suggest that PMCA can faithfully replicate aspects of cross-species transmission potential and might provide useful additional information concerning the molecular barrier to zoonotic transmission.
The establishment of a causative link between BSE and vCJD clearly involved epidemiology, clinical and neuropathological investigation, but as shown above, it also involved the use of carefully selected in vivo and in vitro model systems. The history of BSE/vCJD can be used as a paradigmatic example of how to consider possible links between other animal prion diseases and how to investigate zoonotic potential.
Atypical TSEs
Following the BSE epidemic in the UK, active surveillance programs for animal prion diseases in Europe and elsewhere led to the identification of rare atypical forms of prion diseases in sheep and cattle. These included Nor98 or atypical scrapie in sheepCitation28 and two prion diseases of cattle: bovine amyloidotic spongiform encephalopathy or L-type BSECitation29 and H-type BSE,Citation30 both of which have a pathology and epidemiology distinct from classical (C-type) BSE.
Atypical scrapie: Nor98
First identified in Norway in 1998, Nor98 affected animals presented with progressive ataxia and showed vacuolar lesions and PrPSc accumulation in the cerebral and cerebellar distinct from classical scrapie. Nor98 affected sheep are of Prnp genotypes rarely affected by the classical scrapie. Molecular analysis of protease-resistant prion protein (PrPres) in the brain revealed the presence of a low molecular weight PrPres band hitherto not described in animal TSE. These features suggested the identification of a novel scrapie phenotype or strain.Citation28,Citation31
Transmission of scrapie and atypical scrapie to a transgenic mice model that overexpresses the ovine prion protein of the ARQ/ARQ Prnp genotype showed differences in the transmission rate between these two sheep TSEs, suggesting that they represent different strains and that the risk of transmission of atypical scrapie (between sheep) is lower than that of classical scrapie.Citation32 Recently, Wilson and coworkers reported failure of transmission of Nor98 in humanized transgenic model mice, arguing for a substantial transmission barrier between atypical scrapie and humans.Citation33
L-BSE/ H-BSE: in vivo and in vitro models
Intracranial inoculation of cynomolgus monkeys (Macacca fasicularies) with C-BSE, L-type BSE and vCJD revealed a distinctive pathological profile for L-type BSE. The animals challenged with L-type BSE had a shorter survival time compared with that for the C-BSE inoculated macaques, even although the dose was 4 times less (in terms of the amount of tissue used) compared with the C-BSE inoculum. Furthermore, the C-BSE inoculum was 10-fold times more concentrated in terms of PrPres than the L-BSE material. These results demonstrate the susceptibility of non-human primates to L-BSE and suggest greater pathogenicity of L-BSE than C-BSE to non-human primates.Citation34
Active animal TSE surveillance programs worldwide have identified H-BSE and L-BSE in countries outside the European Union (e.g., Japan). To evaluate the infectivity and the prion disease phenotype of the Japanese L-BSE isolates, cynomolgus macaques were intracranially inoculated with a brain macerate from two confirmed cases of L-BSE. Similar to the report of Comoy and coworkers,Citation34 the incubation periods and the duration of the disease were approximately 2/3 shorter than those of C-BSE, suggesting once again that L-BSE may be more virulent than the C-BSE in non-human primates.Citation35
Following the suggestion that L-type BSE could be more virulent than C-BSE, researchers have made pointed efforts to quantify the risk associated with L-BSE and H-BSE to the human health. Beringue and coworkers reported that humanized PrP 129-M overexpressing mice were susceptible to L-type BSE with 100% attack rate and shorter incubation periods compared with C-BSE. Contrary to this finding, H-BSE failed to transmit in the same mice model. The authors suggested a higher theoretical risk of transmission of L-BSE to humans compared with C-BSE.Citation36
Recently, a full range of atypical animal prion diseases were used to challenge humanized transgenic mice that express physiological levels of the human prion protein.Citation33 In contrast to the findings of Beringue and coworkers, L-BSE and H-BSE failed to show any signs of clinical disease or prion pathology, suggesting a substantial transmission barrier between the atypical forms of BSE and humans.Citation33
To our knowledge, the only evidence comparing L-BSE and H-BSE in a cell free system comes from work by our group. A panel composed of several animal TSEs were tested for their ability to convert the human prion protein by PMCA. Human brain and humanized transgenic mouse brain homogenates of the PRNP codon 129 MM and VV genotypes all failed to support amplification when the PMCA reactions were seeded with L-BSE, H-BSE, scrapie and atypical scrapie. In contrast C-BSE and vCJD PrPSc efficiently converted the human PrPC, albeit in a codon 129 (M allele) dependent manner. These findings suggest that, at least at the molecular level, atypical scrapie and atypical BSE present a lower level of risk of zoonotic disease than classical BSE.Citation37
Chronic Wasting Disease: A Risk As Yet Unquantified?
First recognized as a TSE in the early 1980s by Williams and coworkers, chronic wasting disease (CWD) is a transmissible spongiform encephalopathy that affects North American mule deer (Odocoileus hemionus), white-tailed deer (Odocoileus virginianus), Rocky Mountain elk (Cervus elaphus nelsoni), and less frequently moose (Alces alces shirasi).Citation38 After being first identified in Colorado and Wyoming, the disease has been identified in 15 states across the USA, two Canadian provinces and 15 states and provinces in South Korea .Citation39 Clinical signs in animals affected by CWD include progressive loss of weight, pronounced behavioral changes, excessive salivation, and ataxia with head tremors.
Surveillance for CWD-related disease in human
The possibility of CWD affecting human health will be a function of exposure to the CWD agent (or agents) and of human susceptibility to these agents. Dose and route could reasonably be expected to be relevant aspects of exposure and as with other human prion diseases the PRNP codon 129 polymorphisms may be expected to exert an effect on human susceptibility to CWD. The disseminated nature of PrPSc and infectivity in animals infected with CWD, coupled with the popularity of deer hunting in CWD endemic areas, suggests the most likely form of human exposure to CWD infectivity is by the oral route. CJD surveillance systems in CWD affected countries have been alert to this possibility and have been vigilant for CJD patients with suspicious clinical signs and symptoms, a young age at onset and potential risk factors such as game hunting and venison consumption. The literature contains five such cases.Citation40,Citation41 The results of intensive investigation provided diagnoses of sCJD,Citation40 familial CJD and early-onset Alzheimer diseaseCitation41 and on balance the authors discounted a link to CWD, but conceded the difficulties involved in coming to this conclusion. It is important to note however that the specific features of an animal prion disease need not be conserved when zoonoses occur. Neither the characteristic vCJD neuropathology nor the pronounced peripheral tissue involvements of vCJD are features of BSE in cattle. The absence of neuropathological similarities between CWD and any individual cases of human prion disease is not sufficient grounds to discount CWD as a human pathogen. Continued surveillance and epidemiology are required. In parallel the susceptibility of humans to the CWD agent has been investigated using in vivo and in vitro model systems.
Transmission and bioassays in cervids
CWD is thought to be horizontally transmitted between deer by direct contact (e.g., by saliva) and indirectly through pasture contamination with, urine or faeces (reviewed by Miller et al.).Citation42 Experimental exposure of uninfected mule deer to contaminated excreta and decomposed carcasses (of infected animals) shows that CWD prions can persist in the environment for two years.Citation43 Using bioassays in deer, Mathiason and coworkers evaluated the presence of CWD prions in body fluids and excretions in the preclinical phase of the infection. They also evaluated the transmission of the disease using repeated environmental exposure. Positive results were found. Blood and saliva were detected as a potential source of CWD prions. Additionally, the infectious agent (experimentally shed in the environment) was found to be sufficient to transmit the disease to naive deer.Citation44 These observations underline the risks associated with both direct and indirect transmission between deer.
Non-human primate models
In order to test the transmissibility of CWD to non-human primates, Marsh and coworkers reported the experimental challenge of two squirrel monkeys (Saimiri sciureus) with brain homogenate of CWD infected mule deer. The two animals were euthanized at 31 and 34 months post inoculation. Both animals showed a progressive neurological disease, detection of PrPres by western blot and histopatological spongiform changes in the brain, all consistent with a TSE.Citation45
Based on the evidence that non-human primates are susceptible to CWD infection, Race and coworkers evaluated (1) the susceptibility of cynomolgus macaques (Macaca fasicularis) and squirrel monkeys to CWD infection, (2) the different possible routes of inoculation (intracranial and oral) and (3) various sources of the inoculum (representing wild and captive deer and elk). Intracranial inoculation of squirrel monkeys showed that independent of the origin, 80% of the animals developed signs of a spongiform encephalopathy and accumulated PrPres in the brain. Oral exposure with the infectious agent showed 15% of the squirrel monkeys had the presence of PrPSc in the brain and in peripheral tissue. In contrast, cynomolgus macaque inoculated oraly or intracerebraly failed to show evidence of clinical disease 70 months post inoculation.Citation46 Considering relationship of these two non-human primates species to humans, the authors suggested a pronounced species barrier to CWD in humans.
Transgenic mouse models
Modeling the zoonotic transmission of CWD prions to humans, Kong and coworkers reported the inoculation of humanized transgenic mice 129-M lines (expressing the transgene PrP one and 2-fold compared with physiological levels) with CWD elk brain homogenate. After approximately 700 days none of the transgenic mice showed signs of prion disease, and the authors concluded that a substantial species barrier to transmission must be present.Citation47 Sandberg and coworkers confirmed the previous report of Kong et al., that CWD fails to transmit to transgenic mice, irrespective of whether the mice expressed (1) bovine, ovine or human PrP, (2) the human 129-M or 129-V PrP allelic variants, or (3) whether the CWD isolates were from mule deer, elk or white tailed deer.Citation48,Citation49
In order to evaluate the transmissibility of a range of animal TSEs to transgenic animals that express physiological levels the human prion protein, atypical scrapie, C-BSE, H-BSE, L-BSE and CWD were used to challenge the humanised transgenic mice constructed by gene replacement.Citation33 All TSEs, including the CWD isolate used (derived from white tail deer infected animal), failed to produce disease (or signs of infection) on first experimental passage, whether the mice were homozygous for methionine or valine, or heterozygous at codon 129 of PRNP, again suggesting a substantial species barrier between the atypical TSEs and humans.Citation33
In vitro conversion systems: modeling human PrP susceptibility to conversion by chronic wasting disease
In the earliest attempt to assess molecular barriers of the human prion protein conversion by animal prion diseases, Raymond and coworkers compared the ability of CWD, C-BSE, sheep scrapie, and CJD brain homogenates to convert human prion protein metabolically labeled and purified from transfected cells.Citation26 Limited conversion of human PrP by CWD, C-BSE and scrapie was observed, but the model system was unable to discriminate between the molecular susceptibility of the two PRNP genotypes (129-M and 129-V) used in the conversion process. These results revealed a substantial molecular barrier to conversion of PrPC by CWD, C-BSE and sheep scrapie prions.
Using PMCA, Kurt and coworkers reported failure to support in vitro conversion of the human prion protein (independent of the PRNP 129 M or V polymorphisms) when the reaction was seeded with CWD brain homogenate.Citation50 In contrast, Barria and coworkers observed that in vitro conditioning of a mule deer-CWD isolate by PMCA in cervid substrate (or passage in cervidised mice) allowed for subsequent efficient in vitro amplification in a humanised transgenic mouse substrate.Citation51
We have recently conducted a side-by-side comparison of the zoonotic potential of a wide range of animal prions including BSE, atypical BSE, scrapie, atypical scrapie, and CWD using PMCA.Citation37 We found that, unlike atypical scrapie and H- and L-BSE, the CWD specimen tested was able to effect the conversion of human PrPC. The efficiency of conversion was less than that of C-BSE, but it was detectable whether human brain, humanised transgenic mouse brain or human cell homogenates were used as the PMCA substrate. Both the 129-M and 129-V PRNP polymorphic variants were converted by the CWD brain homogenate, but the conversion of 129-M was found to be the more efficient. The CWD-PMCA reaction product retained the ability to propagate in the human PrPC-containing substrate in a second round of PMCA. The human PMCA reaction product shared features with type 1 PrPres ().Citation37
Figure 2. Human PMCA reactions with CWD prions. (A) CWD PrPSc amplification was conducted with substrates from three different sources (human brain, humanized transgenic mouse brain, human cell line,), each with both the PRNP codon 129 MM and the VV genotypes. The susceptibility of human PrPC to conversion was evaluated after a single round of PMCA. Irrespective to the origin, the three substrates supported amplification after one round (96 cycles) of PMCA. CWD amplification showed a preference for the MM genotype, with a robust amplification, compared with the VV genotype. Amplified sample (+), non-amplified sample (-). (B) PrPres typing of the CWD PrPSc amplified in a human brain substrate. vCJD, sCJD VV2 subtype and sCJD MM1 subtype were used as PrPres type reference standards. The CWD PrPres material produced by PMCA (CWD-Hu PMCA) resemble type 1 human PrPres. PrP detection antibody was 3F4. M, Molecular marker.
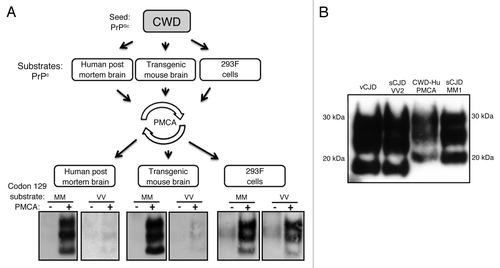
These results would seem to suggest that CWD does indeed have zoonotic potential, at least as judged by the compatibility of CWD prions and their human PrPC target. Furthermore, extrapolation from this simple in vitro assay suggests that if zoonotic CWD occurred, it would most likely effect those of the PRNP codon 129-MM genotype and that the PrPres type would be similar to that found in the most common subtype of sCJD (MM1).
Our study has obvious limitations. First of all, it does not take account of factors operating above the sub-cellular level. Although the prion hypothesis locates the major determinants of prion disease pathogenesis in prion protein structure it is clear that additional factors affect the zoonotic potential of animal prion diseases, such as route of exposure, dose, host genetics, age and co-existing morbidities. A second limitation is that our study was conducted with a single specimen of CWD in an elk. CWD affects several cervid species and in some of these species there exist Prnp polymorphisms and different strains of CWD agent. Before generalizing too broadly from these results, it will be critical to test a wide variety of CWD isolates from different cervid species, polymorphic genotypes and geographical locations to determine whether heterogeneity of zoonotic potential exists within CWD.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
Funding for this research was provided by (1) English Department of Health and the Scottish Government , Grant reference number: 007/0190 and (2)Centre for Clinical Brain Science (CCBS), The University of Edinburgh. UK.
Related Research Data
References
- Prusiner SB. Prions. Proc Natl Acad Sci U S A 1998; 95:13363 - 83; http://dx.doi.org/10.1073/pnas.95.23.13363; PMID: 9811807
- Prusiner SB. Novel proteinaceous infectious particles cause scrapie. Science 1982; 216:136 - 44; http://dx.doi.org/10.1126/science.6801762; PMID: 6801762
- Griffith JS. Self-replication and scrapie. Nature 1967; 215:1043 - 4; http://dx.doi.org/10.1038/2151043a0; PMID: 4964084
- Pattison IH, Jones KM. The possible nature of the transmissible agent of scrapie. Vet Rec 1967; 80:2 - 9; http://dx.doi.org/10.1136/vr.80.1.2; PMID: 4961994
- Oesch B, Westaway D, Wälchli M, McKinley MP, Kent SB, Aebersold R, Barry RA, Tempst P, Teplow DB, Hood LE, et al. A cellular gene encodes scrapie PrP 27-30 protein. Cell 1985; 40:735 - 46; http://dx.doi.org/10.1016/0092-8674(85)90333-2; PMID: 2859120
- Hsiao KK, Scott M, Foster D, Groth DF, DeArmond SJ, Prusiner SB. Spontaneous neurodegeneration in transgenic mice with mutant prion protein. Science 1990; 250:1587 - 90; http://dx.doi.org/10.1126/science.1980379; PMID: 1980379
- Nazor KE, Kuhn F, Seward T, Green M, Zwald D, Pürro M, Schmid J, Biffiger K, Power AM, Oesch B, et al. Immunodetection of disease-associated mutant PrP, which accelerates disease in GSS transgenic mice. EMBO J 2005; 24:2472 - 80; http://dx.doi.org/10.1038/sj.emboj.7600717; PMID: 15962001
- Büeler H, Aguzzi A, Sailer A, Greiner RA, Autenried P, Aguet M, Weissmann C. Mice devoid of PrP are resistant to scrapie. Cell 1993; 73:1339 - 47; http://dx.doi.org/10.1016/0092-8674(93)90360-3; PMID: 8100741
- Aguzzi A, Heikenwalder M, Polymenidou M. Insights into prion strains and neurotoxicity. Nat Rev Mol Cell Biol 2007; 8:552 - 61; http://dx.doi.org/10.1038/nrm2204; PMID: 17585315
- Hill AF, Collinge J. Species-barrier-independent prion replication in apparently resistant species. APMIS 2002; 110:44 - 53; http://dx.doi.org/10.1034/j.1600-0463.2002.100106.x; PMID: 12064255
- Hagiwara K, Hara H, Hanada K. Species-barrier phenomenon in prion transmissibility from a viewpoint of protein science. J Biochem 2013; 153:139 - 45; http://dx.doi.org/10.1093/jb/mvs148; PMID: 23284000
- Kocisko DA, Come JH, Priola SA, Chesebro B, Raymond GJ, Lansbury PT, Caughey B. Cell-free formation of protease-resistant prion protein. Nature 1994; 370:471 - 4; http://dx.doi.org/10.1038/370471a0; PMID: 7913989
- Saborio GP, Permanne B, Soto C. Sensitive detection of pathological prion protein by cyclic amplification of protein misfolding. Nature 2001; 411:810 - 3; http://dx.doi.org/10.1038/35081095; PMID: 11459061
- Castilla J, Gonzalez-Romero D, Saá P, Morales R, De Castro J, Soto C. Crossing the species barrier by PrP(Sc) replication in vitro generates unique infectious prions. Cell 2008; 134:757 - 68; http://dx.doi.org/10.1016/j.cell.2008.07.030; PMID: 18775309
- Smith PG, Bradley R. Bovine spongiform encephalopathy (BSE) and its epidemiology. Br Med Bull 2003; 66:185 - 98; http://dx.doi.org/10.1093/bmb/66.1.185; PMID: 14522859
- Ducrot C, Arnold M, de Koeijer A, Heim D, Calavas D. Review on the epidemiology and dynamics of BSE epidemics. Vet Res 2008; 39:15; http://dx.doi.org/10.1051/vetres:2007053; PMID: 18187031
- Will RG, Ironside JW, Zeidler M, Cousens SN, Estibeiro K, Alperovitch A, Poser S, Pocchiari M, Hofman A, Smith PG. A new variant of Creutzfeldt-Jakob disease in the UK. Lancet 1996; 347:921 - 5; http://dx.doi.org/10.1016/S0140-6736(96)91412-9; PMID: 8598754
- Bishop MT, Pennington C, Heath CA, Will RG, Knight RS. PRNP variation in UK sporadic and variant Creutzfeldt Jakob disease highlights genetic risk factors and a novel non-synonymous polymorphism. BMC Med Genet 2009; 10:146; http://dx.doi.org/10.1186/1471-2350-10-146; PMID: 20035629
- Lasmézas CI, Deslys JP, Demaimay R, Adjou KT, Lamoury F, Dormont D, Robain O, Ironside J, Hauw JJ. BSE transmission to macaques. Nature 1996; 381:743 - 4; http://dx.doi.org/10.1038/381743a0; PMID: 8657276
- Ironside JW. Prion diseases in man. J Pathol 1998; 186:227 - 34; http://dx.doi.org/10.1002/(SICI)1096-9896(1998110)186:3<227::AID-PATH174>3.0.CO;2-3; PMID: 10211109
- Bruce ME, Will RG, Ironside JW, McConnell I, Drummond D, Suttie A, McCardle L, Chree A, Hope J, Birkett C, et al. Transmissions to mice indicate that ‘new variant’ CJD is caused by the BSE agent. Nature 1997; 389:498 - 501; http://dx.doi.org/10.1038/39057; PMID: 9333239
- Scott MR, Will R, Ironside J, Nguyen HO, Tremblay P, DeArmond SJ, Prusiner SB. Compelling transgenetic evidence for transmission of bovine spongiform encephalopathy prions to humans. Proc Natl Acad Sci U S A 1999; 96:15137 - 42; http://dx.doi.org/10.1073/pnas.96.26.15137; PMID: 10611351
- Hill AF, Desbruslais M, Joiner S, Sidle KC, Gowland I, Collinge J, Doey LJ, Lantos P. The same prion strain causes vCJD and BSE. Nature 1997; 389:448 - 50, 526; http://dx.doi.org/10.1038/38925; PMID: 9333232
- Asante EA, Linehan JM, Desbruslais M, Joiner S, Gowland I, Wood AL, Welch J, Hill AF, Lloyd SE, Wadsworth JD, et al. BSE prions propagate as either variant CJD-like or sporadic CJD-like prion strains in transgenic mice expressing human prion protein. EMBO J 2002; 21:6358 - 66; http://dx.doi.org/10.1093/emboj/cdf653; PMID: 12456643
- Bishop MT, Hart P, Aitchison L, Baybutt HN, Plinston C, Thomson V, Tuzi NL, Head MW, Ironside JW, Will RG, et al. Predicting susceptibility and incubation time of human-to-human transmission of vCJD. Lancet Neurol 2006; 5:393 - 8; http://dx.doi.org/10.1016/S1474-4422(06)70413-6; PMID: 16632309
- Raymond GJ, Hope J, Kocisko DA, Priola SA, Raymond LD, Bossers A, Ironside J, Will RG, Chen SG, Petersen RB, et al. Molecular assessment of the potential transmissibilities of BSE and scrapie to humans. Nature 1997; 388:285 - 8; http://dx.doi.org/10.1038/40876; PMID: 9230438
- Jones M, Wight D, Barron R, Jeffrey M, Manson J, Prowse C, Ironside JW, Head MW. Molecular model of prion transmission to humans. Emerg Infect Dis 2009; 15:2013 - 6; http://dx.doi.org/10.3201/eid1512.090194; PMID: 19961689
- Benestad SL, Sarradin P, Thu B, Schönheit J, Tranulis MA, Bratberg B. Cases of scrapie with unusual features in Norway and designation of a new type, Nor98. Vet Rec 2003; 153:202 - 8; http://dx.doi.org/10.1136/vr.153.7.202; PMID: 12956297
- Casalone C, Zanusso G, Acutis P, Ferrari S, Capucci L, Tagliavini F, Monaco S, Caramelli M. Identification of a second bovine amyloidotic spongiform encephalopathy: molecular similarities with sporadic Creutzfeldt-Jakob disease. Proc Natl Acad Sci U S A 2004; 101:3065 - 70; http://dx.doi.org/10.1073/pnas.0305777101; PMID: 14970340
- Biacabe AG, Laplanche JL, Ryder S, Baron T. Distinct molecular phenotypes in bovine prion diseases. EMBO Rep 2004; 5:110 - 5; http://dx.doi.org/10.1038/sj.embor.7400054; PMID: 14710195
- Tranulis MA, Benestad SL, Baron T, Kretzschmar H. Atypical prion diseases in humans and animals. Top Curr Chem 2011; 305:23 - 50; http://dx.doi.org/10.1007/128_2011_161; PMID: 21598097
- Arsac JN, Bétemps D, Morignat E, Féraudet C, Bencsik A, Aubert D, Grassi J, Baron T. Transmissibility of atypical scrapie in ovine transgenic mice: major effects of host prion protein expression and donor prion genotype. PLoS One 2009; 4:e7300; http://dx.doi.org/10.1371/journal.pone.0007300; PMID: 19806224
- Wilson R, Plinston C, Hunter N, Casalone C, Corona C, Tagliavini F, Suardi S, Ruggerone M, Moda F, Graziano S, et al. Chronic wasting disease and atypical forms of bovine spongiform encephalopathy and scrapie are not transmissible to mice expressing wild-type levels of human prion protein. J Gen Virol 2012; 93:1624 - 9; http://dx.doi.org/10.1099/vir.0.042507-0; PMID: 22495232
- Comoy EE, Casalone C, Lescoutra-Etchegaray N, Zanusso G, Freire S, Marcé D, Auvré F, Ruchoux MM, Ferrari S, Monaco S, et al. Atypical BSE (BASE) transmitted from asymptomatic aging cattle to a primate. PLoS One 2008; 3:e3017; http://dx.doi.org/10.1371/journal.pone.0003017; PMID: 18714385
- Ono F, Tase N, Kurosawa A, Hiyaoka A, Ohyama A, Tezuka Y, Wada N, Sato Y, Tobiume M, Hagiwara K, et al. Atypical L-type bovine spongiform encephalopathy (L-BSE) transmission to cynomolgus macaques, a non-human primate. Jpn J Infect Dis 2011; 64:81 - 4; PMID: 21266763
- Béringue V, Herzog L, Reine F, Le Dur A, Casalone C, Vilotte JL, Laude H. Transmission of atypical bovine prions to mice transgenic for human prion protein. Emerg Infect Dis 2008; 14:1898 - 901; http://dx.doi.org/10.3201/eid1412.080941; PMID: 19046515
- Barria MA, Balachandran A, Morita M, Kitamoto T, Barron R, Manson J, Knight R, Ironside JW, Head MW. Molecular barriers to zoonotic transmission of prions. Emerg Infect Dis 2014; 20:88 - 97; http://dx.doi.org/10.3201/eid2001.130858; PMID: 24377702
- Williams ES, Young S. Chronic wasting disease of captive mule deer: a spongiform encephalopathy. J Wildl Dis 1980; 16:89 - 98; http://dx.doi.org/10.7589/0090-3558-16.1.89; PMID: 7373730
- Saunders SE, Bartelt-Hunt SL, Bartz JC. Occurrence, transmission, and zoonotic potential of chronic wasting disease. Emerg Infect Dis 2012; 18:369 - 76; http://dx.doi.org/10.3201/eid1803.110685; PMID: 22377159
- Belay ED, Gambetti P, Schonberger LB, Parchi P, Lyon DR, Capellari S, McQuiston JH, Bradley K, Dowdle G, Crutcher JM, et al. Creutzfeldt-Jakob disease in unusually young patients who consumed venison. Arch Neurol 2001; 58:1673 - 8; http://dx.doi.org/10.1001/archneur.58.10.1673; PMID: 11594928
- Anderson CA, Bosque P, Filley CM, Arciniegas DB, Kleinschmidt-Demasters BK, Pape WJ, Tyler KL. Colorado surveillance program for chronic wasting disease transmission to humans: lessons from 2 highly suspicious but negative cases. Arch Neurol 2007; 64:439 - 41; http://dx.doi.org/10.1001/archneur.64.3.439; PMID: 17353391
- Miller MW, Williams ES. Prion disease: horizontal prion transmission in mule deer. Nature 2003; 425:35 - 6; http://dx.doi.org/10.1038/425035a; PMID: 12955129
- Miller MW, Williams ES, Hobbs NT, Wolfe LL. Environmental sources of prion transmission in mule deer. Emerg Infect Dis 2004; 10:1003 - 6; http://dx.doi.org/10.3201/eid1006.040010; PMID: 15207049
- Mathiason CK, Hays SA, Powers J, Hayes-Klug J, Langenberg J, Dahmes SJ, Osborn DA, Miller KV, Warren RJ, Mason GL, et al. Infectious prions in pre-clinical deer and transmission of chronic wasting disease solely by environmental exposure. PLoS One 2009; 4:e5916; http://dx.doi.org/10.1371/journal.pone.0005916; PMID: 19529769
- Marsh RF, Kincaid AE, Bessen RA, Bartz JC. Interspecies transmission of chronic wasting disease prions to squirrel monkeys (Saimiri sciureus). J Virol 2005; 79:13794 - 6; http://dx.doi.org/10.1128/JVI.79.21.13794-13796.2005; PMID: 16227298
- Race B, Meade-White KD, Miller MW, Barbian KD, Rubenstein R, LaFauci G, Cervenakova L, Favara C, Gardner D, Long D, et al. Susceptibilities of nonhuman primates to chronic wasting disease. Emerg Infect Dis 2009; 15:1366 - 76; http://dx.doi.org/10.3201/eid1509.090253; PMID: 19788803
- Kong Q, Huang S, Zou W, Vanegas D, Wang M, Wu D, Yuan J, Zheng M, Bai H, Deng H, et al. Chronic wasting disease of elk: transmissibility to humans examined by transgenic mouse models. J Neurosci 2005; 25:7944 - 9; http://dx.doi.org/10.1523/JNEUROSCI.2467-05.2005; PMID: 16135751
- Sandberg MK, Al-Doujaily H, Sigurdson CJ, Glatzel M, O’Malley C, Powell C, Asante EA, Linehan JM, Brandner S, Wadsworth JD, et al. Chronic wasting disease prions are not transmissible to transgenic mice overexpressing human prion protein. J Gen Virol 2010; 91:2651 - 7; http://dx.doi.org/10.1099/vir.0.024380-0; PMID: 20610667
- Tamgüney G, Giles K, Bouzamondo-Bernstein E, Bosque PJ, Miller MW, Safar J, DeArmond SJ, Prusiner SB. Transmission of elk and deer prions to transgenic mice. J Virol 2006; 80:9104 - 14; http://dx.doi.org/10.1128/JVI.00098-06; PMID: 16940522
- Kurt TD, Telling GC, Zabel MD, Hoover EA. Trans-species amplification of PrP(CWD) and correlation with rigid loop 170N. Virology 2009; 387:235 - 43; http://dx.doi.org/10.1016/j.virol.2009.02.025; PMID: 19269662
- Barria MA, Telling GC, Gambetti P, Mastrianni JA, Soto C. Generation of a new form of human PrP(Sc) in vitro by interspecies transmission from cervid prions. J Biol Chem 2011; 286:7490 - 5; http://dx.doi.org/10.1074/jbc.M110.198465; PMID: 21209079