
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Aggregation of α-synuclein plays a crucial role in the pathogenesis of synucleinopathies, a group of neurodegenerative diseases including Parkinson disease (PD), dementia with Lewy bodies (DLB), diffuse Lewy body disease (DLBD) and multiple system atrophy (MSA). The common feature of these diseases is a pathological deposition of protein aggregates, known as Lewy bodies (LBs) in the central nervous system. The major component of these aggregates is α-synuclein, a natively unfolded protein, which may undergo dramatic structural changes resulting in the formation of β-sheet rich assemblies. In vitro studies have shown that recombinant α-synuclein protein may polymerize into amyloidogenic fibrils resembling those found in LBs. These aggregates may be uptaken and propagated between cells in a prion-like manner. Here we present the mechanisms and kinetics of α-synuclein aggregation in vitro, as well as crucial factors affecting this process. We also describe how PD-linked α-synuclein mutations and some exogenous factors modulate in vitro aggregation. Furthermore, we present a current knowledge on the mechanisms by which extracellular aggregates may be internalized and propagated between cells, as well as the mechanisms of their toxicity.
Introduction
Human α-synuclein is a natively unfolded 140-amino-acid residue protein widely expressed in neurons, found predominantly in presynaptic terminals.Citation1-Citation3 It was originally identified at the nuclear level, accounting for the designation “syn-nuclein”,Citation4 but subsequently described as a predominantly cytoplasmic protein.Citation5-Citation7 While an ultimate definition of α-synuclein function has not been provided yet, different studies have shown its role in mediating dopamine synthesis, release and reuptake in dopaminergic neurons, as strongly suggested by the protein presynaptic location and its interaction with membranes.Citation8-Citation13 Additional studies reported α-synuclein upregulation in response to oxidative stress and excitotoxicity, suggesting a role for the protein in protecting neuronal cells against damage.Citation14 In particular, α-synuclein may cooperate with the cysteine-string protein-α (CSPα)—a synaptic vesicle protein with co-chaperone activity—in preventing neurodegeneration.Citation15
Structurally, α-synuclein features an N-terminal domain (residues 1–60), a central hydrophobic portion denoted as non-Aβ component of Alzheimer diseases amyloid (NAC, residues 61–95) and a C-terminal negatively charged region (residues 96–140). Both the N-terminal and NAC domains contain six highly conserved hexameric (KTKEGV) motifs ().Citation16 In its native monomeric state, α-synuclein behaves as an intrinsically unstructured protein, characterized by the lack of a stable tertiary structure.Citation17,Citation18 However, it may adopt a secondary structure upon binding to protein interactors or lipid membranes. Early studies reported that the N-terminal domain of α-synuclein displays α-helical folding in the presence of artificial membranes such as sodium dodecyl sulfate micelles.Citation19-Citation22 Solution state NMR studies on the structures of micelle-bound α-synuclein have shown that the protein contains two distinct α-helices in the region encompassing residues 1–92, while the C-terminal results predominantly unstructured ().Citation23,Citation24 Other studies have attempted to obtain structural information about α-synuclein segments complexed with physiological partners such as calmodulinCitation25 or synphilin-1,Citation26 with a nanobodyCitation27 or the maltose binding proteinCitation28 (). A recent work has reported that natively cell-extracted α-synuclein may be present as a tetramer formed by α-helical folded monomers with no binding to lipid membranes,Citation29 but a further work has shown that α-synuclein in mammalian cells exists as unfolded monomer when dissociated from lipid membranes.Citation30
Figure 1. Structural features of monomeric α-synuclein. Primary amino acidic sequence (UniProtKB: P37840) of human α-synuclein with the imperfect exapeptide repeatsCitation16 underlined (A) and schematic representation of the biochemically different domains including the currently identified PD-related mutations (B). Currently available NMR and X-ray crystal structures of α-synuclein segments (in red) bound to micelles (C and D) PDB: 1XQ8 and 2KKW, respectively, calmodulin, CaM (E) PDB: 2M55, synphylin-1 (F) PDB: 2JN5, nanobody, NbSyn2 (G) PDB: 2X6M and maltose binding protein (H) PDB: 3Q25, 2Q26, 3Q27.
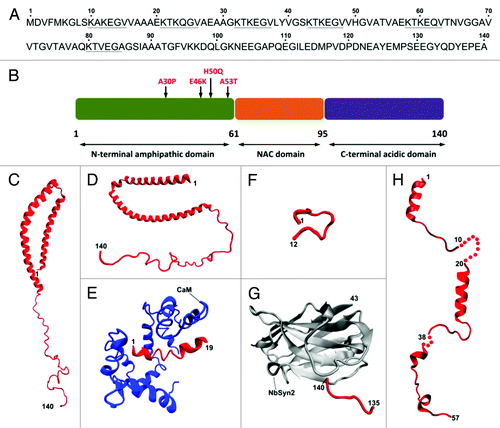
Overall the structural studies on the α-synuclein monomer suggest a model where the lack of secondary structure motifs is the first event leading to its misfolding and aggregation in pathological and disease-related amyloid assemblies found in Parkinson disease (PD) and other synucleinopathies.
In the next sections, we present an overview of the strategies currently used to produce synthetic α-synuclein assemblies and their possible implications for the future of PD research.
In Vitro Aggregation Assays of α-Synuclein
Deposition of Lewy bodies (LBs) and Lewy neurites (LNs) in the brain is a major pathological hallmark of several neurodegenerative diseases, including PD, dementia with Lewy bodies (DLB), diffuse Lewy body disease (DLBD), and multiple system atrophy (MSA). LBs are spherical cytoplasmic inclusions, 5–25 µm in diameter, formed by aggregated proteins, with a dense eosinophilic core and a clearer surrounding halo. The major component of the proteinaceous filaments of LBs and LNs is α-synuclein.Citation31
In vitro studies have revealed that recombinant α-synuclein can assemble into fibrils with morphologies and staining characteristics similar to those extracted from disease-affected brains. Wild-type (WT) α-synuclein fibrils formed in vitro are 8–10 nm in height as determined by atomic force microscopy (AFM) and about 10 nm in width (electron microscopy, EM), while fibrils of PD LBs observed in tissue slices are reported to be about 10 nm in width.Citation32-Citation34 Moreover, the presence of “twisted” fibrils as well as some small filaments of 5 nm width is a common feature of fibrils extracted from brains and some fibrils formed in vitro.Citation31,Citation32
Aggregation of α-synuclein plays a critical role in PD and related neurodegenerative disorders; hence in vitro aggregation assays are powerful molecular tools for studying the mechanisms of neurodegeneration. Understanding the mechanisms by which a soluble α-synuclein is transformed into insoluble β-rich fibrils, as well as describing the structural features of these fibrils are still questions to be addressed in future research. Additionally, α-synuclein aggregation assays may be feasible tools for the development of new drug compounds capable of interfering with α-synuclein fibril formation. Increasing evidence demonstrates that in vitro formed fibrils are internalized by cells and may be propagated in a prion-like manner leading to cytotoxicity. Thus, in vitro aggregation of α-synuclein may yield crucial components for studying molecular mechanisms of aggregate uptake, spreading and toxicity.
Strategies for the purification of recombinant α-synuclein
In vitro fibrillization assays require high amounts of highly pure α-synuclein protein. Recombinant α-synuclein is usually produced in E. coli cells and purified from either whole cell extract or periplasmic extract. Traditionally, the protein is purified from the whole cell extract by sonication or boiling, or ammonium sulfate precipitation followed by anion exchange and size exclusion chromatography purifications.Citation35-Citation37 Ren and collaborators have shown that E.coli-expressed α-synuclein without signal sequence mostly localizes in the bacterial periplasm.Citation38 The protein translocation appeared to be mediated by the signal recognition particle (SRP)-dependent pathway facilitated by the α-synuclein C-terminal domain (residues 90–140) as a major part responsible for the translocation.Citation38 Therefore, a new efficient and fast method has been developed for α-synuclein extraction and purification from periplasm.Citation39 Briefly, the protein is first extracted from bacterial periplasm by osmotic shock, followed by anion-exchange chromatography purification. One L of E.coli LB medium culture yields about 80 mg of highly pure α-synuclein.Citation39
Tools for monitoring α-synuclein fibril formation in vitro
In vitro fibrillization assays require powerful tools for detecting fibril formation and monitoring the fibrillization kinetics in real time. Two common histological dyes, Thioflavin T (ThT) and Congo red (CR), are used to this end. The mechanism of interaction between both dyes and amyloid fibrils remains poorly understood; both compounds seem nevertheless to interact with the cross-β structures, exhibiting changes in fluorescence. ThT is a benzothiazole dye that exhibits enhanced fluorescence upon binding to amyloid fibrils and constitutes the mostly used extrinsic probe for monitoring fibrillization kinetics in vitro. In the presence of fibrils, ThT gives rise to an excitation maximum of 450 nm and enhanced emission at 482 nm, whereas unbound ThT is not fluorescent at these wavelengths.Citation40 ThT is a reliable and specific fibril-detecting tool, which does not interfere with the kinetics of α-synuclein fibrillization.Citation41 However, despite its specificity toward fibrils, a slight increase in ThT fluorescence has been detected after binding to amorphous α-synuclein aggregates.Citation42 The use of ThT for monitoring fibril formation should be optimized and negative controls are needed, as spectral properties of this dye may vary under different conditions. Additionally, being ThT a charged and hydrophobic molecule, it may non-specifically bind to some proteins by electrostatic and hydrophobic interactions.Citation43
CR seems unsuitable for monitoring α-synuclein aggregation, since it exhibits high affinity binding to α-synuclein and has an inhibitory effect on the fibrillization assay.Citation44 Some studies have indicated that CR may interfere with protein misfolding and aggregation by stabilizing native protein monomers or partially folded intermediates.Citation45
Since the sensitivity and specificity of ThT and CR have been questioned, several efforts have been made to design extrinsic probes and spectroscopic techniques for monitoring the fibrillization process. Celej and coworkers reported that N-arylaminonaphthalenesulfonate (NAS) and its derivatives act as non-covalent, highly sensitive probes of α-synuclein fibril formation.Citation46 Recently, it has been shown that protein aggregation reactions can be spectroscopically monitored without the need for extrinsic labels. Some studies have reported that amyloid-like structure is associated with an intrinsic fluorescent phenomenon that occurs in the visible range.Citation47-Citation49 The physical mechanism underlying the intrinsic fluorescence emission remains unknown. It has been suggested that the hydrogen-bonded water molecules within the cross-β structure might play a key role in the fluorescence emission of the fibrils. Direct excitation of the fibrils could induce electronic transitions in peptides because of a partial delocalization of peptide electrons elicited by the presence of hydrogen bonds.Citation47 Pinotsi and collaborators have shown that this phenomenon can be used for monitoring the aggregation of proteins with no need for extrinsic labels, thus avoiding potential interference with the aggregation process.Citation50 This method appears feasible and reproducible; it requires a confocal microscope and it has been successfully used for monitoring α-synuclein fibrillization in vitro.Citation50
Mechanism and kinetics of fibril formation in vitro
During the fibrillization process, a soluble protein is converted into insoluble highly ordered aggregates exhibiting β-sheet enriched structures. Fibrillization of globular proteins requires the presence of partially unfolded conformations in order to adopt a structure competent for self-assembly into fibrils.Citation51 Partial unfolding exposes the hydrophobic regions of the protein, thus facilitating the interactions between exposed amyloidogenic regions of two proteins and subsequent formation of higher order species. Several factors destabilizing the native fold of the protein, such as mutations, denaturants, changes in pH, or elevated temperature, have been shown to enhance fibril formation.Citation51
Fibrillization of natively unfolded proteins such as α-synuclein presents intriguing issues, since it remains unclear how an intrinsically unstructured protein may form highly organized β-structured fibrils. Partial folding of the protein seems to represent a crucial step in the fibrillization pathway.Citation51 Natively unfolded proteins are characterized by low hydrophobicity and a large net charge.Citation52 It is supposed that any factors increasing protein hydrophobicity or decreasing protein net charge may lead to its partial folding and thus promote the fibrillogenic pathway. The overall hydrophobicity of a protein increases with higher temperature.Citation53 It has been reported that either a decrease in pH or an increase in temperature leads α-synuclein to adopt a partially folded conformation, a change correlated with the enhanced formation of fibrils.Citation41
The model for α-synuclein fibrillization has been proposed as follows:
U ↔ I ↔ nucleus → F,
where U corresponds to natively unfolded conformation, I: partially folded intermediate, nucleus: fibril nucleus, and F: fibril. This model describes the two key kinetic steps in the fibrillization process. The first step is the conformational transformation of α-synuclein into the aggregation-competent, partially folded intermediate; the second is the formation of the assembly-competent oligomers (nuclei), which is followed by oligomer growth and fibril formation. The model suggests that any factor that promotes intermediate formation (for example point mutations, non-polar molecules, cations, and others) will also promote α-synuclein fibrillization.Citation41
The kinetics of α-synuclein fibrillization is commonly described as a sigmoidal curve, composed of an initial lag phase (nucleation) when the amount of fibrils is too low to be detected, a subsequent exponential phase (elongation) when fibril concentration increases rapidly, and a final plateau phase indicating the end of fibril formation. The length of lag phase and the fibril growth rates depend on several factors, including initial protein concentration, pH, temperature, agitation and other factors that will be discussed below.
Moreover, the fibrillization process can be catalyzed by adding preformed fibrils (seeds), which act as nuclei.Citation54-Citation56 This effect has been termed “‘nucleation-dependent’ fibrillization.” The added seeds may act as catalytic sites that induce conformational changes in α-synuclein and accelerate the reaction rates.Citation54
To compare the effects of different reaction conditions on fibrillization kinetics, ThT fluorescence is commonly plotted as a function of time and fitted to a sigmoidal curve using the equation:
where Y is the fluorescence intensity, x is the time, and x0 is the time to 50% of maximal fluorescence. Thus, the apparent rate constant, kapp, for fibril growth is given by 1/τ and the lag time is given by x0–2τ.Citation57
To test the effects of different factors on fibrillization kinetics, usually the lag phase, the apparent rate constant and/or the maximum intensity of ThT emission intensity at the end of fibril formation are calculated. These values obtained from tested samples are compared using commonly available statistic tests.Citation58 The fibrillization kinetics has been shown to be fairly similar among different amyloidogenic proteins. However, the kinetic profiles do not display always the typical sigmoidal shape since multiple steps may be present, thus affecting the interpretation of the correct lag phase and the apparent rate constant. Optimizing the reproducibility of protein fibrillization is crucial to obtain reliable results.Citation58
Various data confirmed that in vitro aggregation of α-synuclein may yield different final products, such as fibrils, soluble oligomers, or insoluble “amorphous aggregates.” Aggregation assays resulted in the formation of several oligomeric species with different morphologies, including spherical, chain-like, and annular oligomers.Citation59 However, the mechanism of conversion among different oligomers is not fully understood.
The generation of different α-synuclein conformations has also been associated with the pathogenesis of PD and other synucleinopathies; fibrillar forms seem nevertheless to be the main component of LBs.Citation31
Structural features of human α-synuclein fibrils
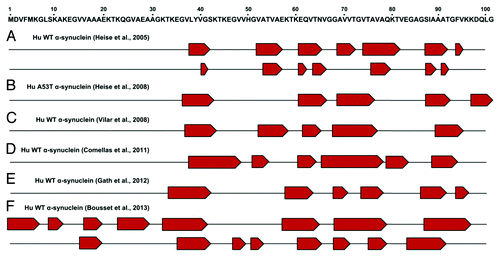
The assembly of physiologically expressed proteins into oligomeric and fibrillar aggregates with cross-β structure is an essential event of different human neurodegenerative diseases, such as PD, Alzheimer disease, and prion diseases. It is generally accepted that aggregation takes place from partially structured states of proteins. However, the role of the residual structures present in such conformational states is not well understood. α-synuclein fibrillar aggregates are pathological hallmarks of PD and other related synucleinopathies. The structural organization of α-synuclein amyloid fibrils has been researched extensively. Amyloid fibrils are composed of several protofilaments arranged into the classical cross-β conformation, with individual β-strands running perpendicular to the fiber axis.Citation34,Citation60,Citation61 Findings from electron paramagnetic resonance (EPR)Citation62,Citation63 and magic-angle spinning solid-state NMR (ssNMR) have shown that the amyloidogenic β-sheet core is mostly located within the central NAC domain.Citation64-Citation71 illustrates an updated view of the β-sheet organization into the α-synuclein amyloid fibrils as determined by ssNMR. Although the fibril core spans approximately residues 38 to 100, structural variations of local β-strands have been reported inside this region. A recent report has expanded current knowledge of the α-synuclein segments involved in amyloid structural reorganization. In particular, Bousset and collaborators identified two novel α-synuclein assemblies characterized by ordered β-sheet organization starting from residue 1 to 38; this region appeared unstructured in all previous ssNMR studies.Citation65 Interestingly, the same group has reported that the two α-synuclein polymorphs showed marked differences in their biological functions. Overall the EPR and ssNMR studies highlight the remarkable conformational plasticity of α-synuclein, which can adopt different monomeric, oligomeric, or fibrillar assemblies depending on the environment.
Figure 2. α-synuclein fibril core regions as determined by ssNMR studies. Upper panel: schematic representation of the α-synuclein segment from residue 1 to 100 involved in structural changes. Lower panel: proposed β-strand (indicated as red pentagons) organization of different human (Hu) α-synuclein fibrillar assemblies determined by different groups: (A) reference Citation68 (B) reference Citation67 (C) reference Citation69 (D) reference Citation70 (E) reference Citation71 (F) reference Citation65.
Factors affecting the kinetics of α-synuclein fibrillization in vitro
Oligomers and fibrils produced in vitro under diverse conditions may result in different structural properties and consequently exert different biological functions.Citation72 As mentioned above, several factors including the initial concentration of monomeric protein, molecular crowding, agitation, pH, temperature, and ionic strength, have been shown to greatly affect the kinetics of α-synuclein fibrillization.Citation32,Citation41,Citation73-Citation75 Thus, developing a reliable and reproducible aggregation assay is necessary to screen the effects of exogenous factors on the fibrillization process, as well as to produce different aggregate species for structural studies and test their biological functions. Here, we characterize crucial factors that determine the kinetics of fibril formation in vitro.
α-synuclein concentration
Initial concentration of monomeric α-synuclein is a crucial factor determining the kinetics of fibril formation. Commonly, 0.5 to 1 mg/mL (35–70 µM) of α-synuclein is used during fibrillization assays.Citation39,Citation74,Citation76,Citation77 Other studies have used higher concentrations, namely 5 to 7 mg/mL.Citation78,Citation79 The minimal α-synuclein concentration, below which fibrils are not formed, has been estimated to be approximately 0.2 mg/mL.Citation80 The kinetics of fibril formation is a concentration-dependent process; however, the concentration dependence almost disappears in the presence of higher protein concentrations. Above the supercritical concentration, nucleation is no longer rate-limiting and the fibril growth rate is almost independent of the protein concentration.Citation81 In fact, increasing α-synuclein concentration has been reported to decrease the lag-phase of fibrillization.Citation82 Supercritical concentration of α-synuclein has been estimated to be about 8.2 mg/ml (̴ 574 µM); however this may vary according to the conditions used for the fibrillization reaction.Citation42
Molecular crowding
Macromolecular crowding is another factor significantly affecting aggregation, as it may dramatically accelerate α-synuclein fibril formation.Citation74,Citation83 The effect of molecular crowding on protein aggregation in vitro has been examined by using concentrated solutions of a model “crowding agent” such as polyethylene glycols (PEG), polysaccharides (dextrans or Ficolls), or proteins (for example, lysozyme and bovine serum albumin). All these molecules accelerated α-synuclein fibrillization in vitro.Citation83 The crowding agents had little effect on the conformation of α-synuclein, as shown by far-UV CD spectroscopy.Citation74 It seems that the mechanism by which crowding agents affect fibrillization depends on their concentrations. It has been suggested that at lower crowding agent concentrations, the major effect of macromolecular crowding on protein aggregation is due to excluded volume effect, which favors self-association of α-synuclein following the increase of the effective protein concentration. At a higher concentration, viscosity effects of the polymer solutions may become dominant.Citation74
Agitation
As reported by several studies, agitation is yet another crucial factor impacting the kinetics of the fibrillization process. Agitation of α-synuclein solutions leads to a dramatic increase in the rate of fibrillization, shortening the lag phase from weeks to hours.Citation32,Citation84 Uversky and collaborators have reported that vigorous agitation was the major determinant of the fibrillization rate. Very vigorous agitation attenuated the effects of ionic strength, protein concentration and various additives on the rate of fibril formation.Citation85 The exact mechanism by which agitation induces fibrillization is unknown, however it could increase the air-water interface, thus leading to a partial folding of α-synuclein and the formation of the fibrillization intermediate.Citation82 Moreover, agitation may increase the number of fibril ends through fragmentation or increase the collision of monomeric and oligomeric species interacting with fibril ends.Citation86
Agitation is commonly performed either in a glass vial with a mini stir bar or in microplates on a fluorescence plate reader. With the former method, termed single time-point dilution ThT assay,Citation39,Citation41,Citation75,Citation87,Citation88 aliquots are taken out at given time intervals for ThT fluorescence measurement, whereas in the latter approach (in situ real-time assay) ThT fluorescence is monitored in real time at regular intervals.Citation58,Citation73,Citation74 Agitation speed in different assays ranges from 120 rpmCitation89 to 1000 rpm.Citation78 Agitation with glass beads has been shown to significantly decrease the lag time of the fibrillization and to increase the reproducibility of the assay.Citation58
Reproducibility is an important issue in in vitro fibrillization assays, as it may vary significantly across sample replicates and among experiments.Citation58 Giehm and Otzen have tested factors that influence the reproducibility of the fibrillization of α-synuclein in plate reader assays. The authors have determined that increased reproducibility is correlated with decreased lag time. Orbital agitation using a small glass bead in each well significantly increased lag time and was crucial to obtain good reproducibility.Citation58
Temperature, pH and ionic strength
Several data have indicated that decreasing the pH or increasing the temperature substantially accelerated the kinetics of α-synuclein fibrillization, affecting both the lag time and the elongation rate. Elevated temperatures have been shown to strengthen hydrophobic interactions whereas lower pH decreased the net charge of the protein.Citation41,Citation53 Both factors induced some structural changes in α-synuclein, as shown by far-UV CD and FTIR spectroscopies, and consequently contributed to a reversible transformation of α-synuclein into a partially folded intermediate, whose formation is crucial for the fibrillization process.Citation41,Citation75
The rate of fibril formation has been shown to increase dramatically with increasing ionic strength, reaching the maximal effect by 300 mM NaCl. Anions have been suggested to induce partial folding of α-synuclein at neutral pH, thus increasing the population of the amyloidogenic intermediate. CD spectra confirmed that at 20 mM concentration, different salts induced partial folding of α-synuclein.Citation73
Exogenous factors modulating α-synuclein fibrillization in vitro
Although the specific etiology of PD is unknown, several factors, both genetic and environmental, may increase the risk of developing the disease.Citation90 In vitro aggregation assays are easy, fast, and powerful tools for screening potential factors that may affect α-synuclein aggregation and thus contribute to PD pathogenesis. Several studies have reported that α-synuclein aggregation may be triggered by environmental and other exogenous factors, such as pesticides, heavy metals, polycations, organic solvents, and some small chemical compounds.Citation91,Citation92 Here, we briefly characterize factors that have been shown either to accelerate or inhibit α-synuclein aggregation in vitro. summarizes exogenous factors that have been involved in the modulation of α-synuclein aggregation.
Table 1. Exogenous factors affecting α-synuclein fibril formation in vitro
Pesticides and herbicides
Uversky and collaborators have reported that certain pesticides and herbicides (rotenone, dieldrin, and paraquat) dramatically accelerated the rate of fibril formation in vitro.Citation85 It has been suggested that the relatively hydrophobic pesticides may bind to the partially folded α-synuclein intermediate, induce its conformational changes and consequently lead to fibril formation.Citation85 In fact, epidemiological studies have shown increased risk of PD in populations exposed to pesticides.Citation93,Citation94
Polycations
Polycations, such as spermine, polylysine, polyarginine, and polyethylenimine, dramatically accelerated α-synuclein fibrillization in vitro. α-synuclein-polycation complex formation was not accompanied by significant structural changes in α-synuclein.Citation77 Electrostatic interactions between polycations and α-synuclein have been suggested to lead to neutralization of the protein net negative charge and to the partially folded amyloidogenic intermediate that initiates fibril formation.Citation77
Metal ions
Several works have described how some metal ions affected α-synuclein fibrillization in vitro. Di- and trivalent ions (e.g., copper, iron, cobalt, and manganese) accelerated the rate of fibril formation. Metal ions stimulated α-synuclein conformational change and led to the development of a partially folded intermediate, a possible critical precursor of fibrils.Citation76,Citation95 Metal exposure has been suggested as one of the factors that may increase the risk of PD. Several epidemiological studies have shown that exposure to some metals, including manganese, iron, aluminum, and copper, was associated with a higher risk of PD.Citation96-Citation99 Moreover, elevated levels of iron, zinc, and aluminum have been reported in the substantia nigra of PD patients, thus confirming their involvement in the pathogenesis.Citation100-Citation102
Histones
Some histones have been shown to form a tight complex with α-synuclein and accelerate its fibrillization in a dose-dependent manner.Citation103 This effect may be physiologically important, as the translocation of α-synuclein into the nucleus and its binding to histones may play a large role in α-synuclein pathology.
Small chemical compounds
Many therapeutic strategies for PD aim at developing chemical compounds and small molecules capable of inhibiting fibril formation. Masuda and collaborators have tested 79 small compounds belonging to different chemical classes (including polyphenols, benzothiazoles, phenothiazoles, steroids, Congo red and its derivatives) for their ability to inhibit the assembly of α-synuclein into filaments in vitro.Citation44 Several of these compounds inhibited α-synuclein assembly; among them, polyphenols proved to be an especially effective class of anti-aggregation compounds. The mechanism underlying the polyphenol-induced inhibition is unclear, but these compounds seem to stabilize soluble, prefibrillar intermediates.Citation44 Meng and coworkers have tested 48 compounds representative of different classes of flavonoids for their ability to inhibit α-synuclein fibril formation in vitro. A variety of flavonoids inhibited α-synuclein in vitro fibrillization to varying extents: some of them (myricetin, tricetin, 6-HP and baicalein) completely inhibited fibril formation and many were able to disaggregate preformed fibrils.Citation104
Heat shock proteins
Heat shock proteins (Hsp) have been reported to interact with α-synuclein and inhibit its aggregation in vitro.Citation105-Citation108 The small Hsp αB-crystallin, a molecular chaperone, inhibited α-synuclein fibrillization. The interaction of α-synuclein with αB-crystallin resulted in large irregular aggregates, suggesting that the chaperone protein redirected α-synuclein from a fibril-formation pathway toward an amorphous aggregation pathway.Citation105 Hsp70 protein inhibited α-synuclein filament formation by binding to soluble prefibrillar intermediates.Citation106,Citation107 Moreover, different Hsp proteins (HspB8, Hsp27, HspB2B3, and αB-crystallin) inhibited the α-synuclein aggregation process, as shown by ThT assay and AFM imaging.Citation108
PAMAM dendrimers
Another class of compounds exerting anti-aggregation effects includes polyamidoamine (PAMAM) dendrimers—highly branched polymers possessing a well-defined spherical geometry.Citation109 PAMAM dendrimers (generations G3, G4, and G5) inhibit fibrillization of α-synuclein and promote the breakdown of pre-existing fibrils. They have been shown to induce structural changes in α-synuclein and redirect the protein to an amorphous aggregation pathway.
Organic solvents
Some factors affecting α-synuclein fibril formation have been reported to inhibit or accelerate the fibrillization depending on their concentration or reaction conditions. Munishkina and collaborators have tested the effect of different organic solvents on α-synuclein fibrillization kinetics.Citation73 Far-UV CD, FTIR and MS spectra confirmed that they induced folding of α-synuclein.Citation73,Citation95 The concentration and composition of different alcohols (ethanol, methanol, and propanol) differently affected the fibrillization rate of α-synuclein.Citation73 Notably, recent studies have established that overexpression of α-synuclein and the consequent higher risk for developing PD is modulated by chronic long-term alcohol exposure.Citation110,Citation111
Effect of PD-related mutations on α-synuclein fibrillization in vitro
Three autosomal dominant missense point mutations in the SNCA gene have been identified in early-onset PD: A53T, A30P, and E46K.Citation112-Citation114 Recently, two independent groups have discovered the novel pathogenic mutation H50Q.Citation115,Citation116 Several works have shown that PD-linked mutations A53T, A46K, and H40Q promoted α-synuclein aggregation in vitro, yet neither of them affected the overall structure of the α-synuclein monomer.Citation32,Citation75,Citation115-Citation120
Mutations A53T and E46K have been shown to significantly accelerate fibrillization in vitro when compared with WT protein.Citation117 Although the A53T and E46K mutated α-synuclein fibrils exhibited distinct morphologies, the rate of amyloid formation of both mutants was similar.Citation121,Citation122 A53T fibrils were about 5–14 nm in width and appeared twisted, with a crossover spacing of about 100 nm; E46K fibrils had often a twisted appearance too, with width varying between about 5 and 14 nm, but with shorter crossover spacing of about 43 nm.Citation121 Recently, Lemkau and collaborators have found by ssNMR that both mutated fibrils possessed site-specific perturbations in their secondary structures, which could alter the overall structural arrangement of the fibril.Citation123
Similarly to WT α-synuclein protein, both mutants generated also prefibrillar intermediates with pore-like activity.Citation60,Citation118,Citation124
The effect of the A30P mutation on fibrillization is not clear, as some data reported that it formed fibrils slightly faster than WT protein,Citation37 whereas others showed that this mutation decreased fibril formation.Citation60,Citation125 Conway and collaborators have observed that A30P had a slower propensity to form fibrils than WT, and it preferentially adopted a soluble, protofibrillar conformation, whereas the WT protein readily progressed to mature fibrils.Citation60 In spite of that, some data have shown that A30P fibril morphologies were similar to those formed from WT protein.Citation34,Citation121 Both WT and A30P fibrils had a filament width of 6–9 nm, which varied slightly along the length of the filaments.Citation121
The recently identified H50Q mutant has been shown to aggregate more readily than WT protein, but more slowly than A53T and E46K.Citation120 AFM studies have shown that, similarly to WT protein, the H50Q protein oligomerized through small oligomers, from protofibrils to mature fibrils.Citation120
Current Knowledge on Internalization and Propagation of α-Synuclein Aggregates
Increasing evidence argues that α-synuclein may transfer from cell to cell, suggesting that prion-like propagation may mediate the pathological progression of PD. Braak and colleagues have proposed that α-synuclein pathology begins in the lower brainstem and olfactory bulb and spreads into the limbic system and eventually to the cortex.Citation126 Two independent studies have revealed host-to-graft propagation of α-synuclein-positive Lewy-like pathology, providing further evidence for prion-like spreading of the pathology.Citation127,Citation128 Embryonic mesencephalic neurons grafted into the neostriatum of PD patients have been shown to accumulate LB pathology. Moreover, α-synuclein was transmitted via endocytosis to neighboring neurons, forming Lewy-like inclusions. α-synuclein was also transmitted from the affected neurons to engrafted neuronal precursor cells in a transgenic (Tg) model of PD-like pathology.Citation129
Several recent studies investigated whether α-synuclein aggregates formed in vitro from recombinant protein may be internalized by cells and eventually recruit endogenous α-synuclein to form LB-like pathology. Increasing evidence suggests that misfolded α-synuclein aggregates, both fibrils and oligomers, may be uptaken and propagated among cells in a prion-like manner and induce pathology in healthy cells.Citation78,Citation79,Citation129,Citation130
Luk and collaborators have shown that addition of in vitro preformed α-synuclein fibrils into culture medium induces intracellular α-synuclein aggregation in different cell lines overexpressing this protein.Citation78 The fibrils were shown to rapidly recruit endogenous soluble protein converting this into detergent-insoluble inclusions, while monomeric and oligomeric α-synuclein showed only a negligible effect. Moreover, α-synuclein inclusions undergo several modifications characteristic for LBs, such as hyperphosphorylation, ubiquitination and accumulation of cytoplasmic vesicles around the periphery of the inclusions.Citation78 Morphological and biochemical similarities between intracellular inclusions and LBs suggest that fibrillar seeds may have a central role in the initial formation of LBs and other disease-associated inclusions. Luk et al. also tested whether exogenous aggregates could induce aggregation of endogenous α-synuclein expressed by primary cells not overexpressing the protein.Citation131 In fact, preformed fibrils entered primary hippocampal neurons and promoted recruitment of soluble α-synuclein into insoluble LB-like aggregates. Consistent with previous results, endogenous aggregates were hyperphosphorylated and ubiquitinated, confirming their LB-like properties. Moreover, formation of endogenous aggregates resulted in the decrease of some synaptic proteins, impairments in neuronal excitability and connectivity and, eventually, neuron loss.Citation131
Another study has shown that intracerebral inoculations of symptomatic mouse brain lysates or preformed α-synuclein fibrils into Tg asymptomatic mice overexpressing the human A53T mutant triggered PD-like pathology and dramatically reduced the survival time of animals.Citation132 PD-like pathology was shown to spread to distal CNS regions. The most severe α-synuclein pathology was detected in brain regions that project to, or receive input from the inoculation sites, suggesting that the propagation of pathology occurs between associated neuronal populations.Citation132 Furthermore, intrastriatal inoculation of synthetic α-synuclein fibrils into wild-type non-Tg mice also led to cell-to-cell transmission of pathologic α-synuclein and LB-like pathology. The α-synuclein pathology caused progressive loss of dopamine neurons in the substantia nigra pars compacta and impairment in motor coordination.Citation130
Injections of in vitro preformed human or mouse α-synuclein fibrils in the substantia nigra of non-Tg mice have also induced LB-like pathology, while mice injected with soluble α-synuclein did not exhibit pathological symptoms.Citation133 Exogenously injected fibrils could be detected for less than one week, but induction of α-synuclein pathology was observed after three months. The pathology induced by exogenous fibrils was similar to that triggered by intracerebral injection of sarcosyl-insoluble α-synuclein from brains of patients with DLB.Citation79
Sacino and collaborators have obtained similar results.Citation134 They have observed that neonatal cerebral injections of exogenous α-synuclein fibrils induced pathology after prolonged incubation times. The injected α-synuclein was rapidly cleared within a few days and inclusion pathology, which arose from endogenous protein, was developed after months. Tg mice overexpressing α-synuclein were more prone to develop pathology. Moreover, the authors have shown that injections of the non-amyloidogenic form of α-synuclein (Δ71–82) induced similar pathology, suggesting that the pathogenesis might be induced not solely by conformation-dependent templating events.Citation134
Altogether these data strongly confirm that exogenous α-synuclein amyloid fibrils produced from recombinant protein have prion-like properties, as they are sufficient to seed and induce pathology in vivo.
A typical hallmark of prion diseases is the existence of different “strains,” arising from the same proteins but having different incubation periods, PrPSc distribution and lesion profiles in the brain, as well as different protease cleavage patterns.Citation135 The existence of different strains has not been identified so far for other neurodegenerative diseases. Recently, Bousset and collaborators have generated in vitro two different high-molecule assemblies of α-synuclein, prepared in buffers with different salt concentrations.Citation65 These assemblies, designated fibrils and ribbons, adopted different conformations, as confirmed by transmission electron microscopy (TEM), FTIR, X-ray diffraction, and ssNMR. Moreover, proteinase-K digestion revealed different cleavage profiles. Fibrils and ribbons exhibited marked differences in their propensity to bind and penetrate cells, toxicity and seeded aggregation of endogenous α-synuclein in vivo. Moreover, they imprinted their intrinsic structure to endogenous α-synuclein, suggesting that different strain-like assemblies may account for the development and progression of different synucleinopathies.Citation65
The mechanism by which exogenous aggregates may be internalized by cells is not clear. Previous studies on fibrils comprised of polyglutamine repeats and tau proteins have shown that exogenous fibrils can be internalized by cells by simple addition into culture medium.Citation136,Citation137 Some studies have reported that a similar internalization process occurs also for α-synuclein,Citation72,Citation129,Citation138 while others have shown that fibrils were efficiently introduced into cells only in the presence of a transduction reagent.Citation56,Citation78 Transduction was performed using cationic-liposomal protein transduction reagent or lipofectamine reagent.Citation56,Citation139
Thus the ability of α-synuclein aggregates to enter into cells and induce aggregation of endogenous protein may depend on the precise structure of the aggregated protein. Danzer and collaborators have observed that, depending on in vitro aggregation conditions, α-synuclein oligomers formed distinct populations that differed in their biophysical properties and cellular effects.Citation72,Citation140 Only some types of oligomers were internalized by primary neuronal cells and neuronal cell lines, and induced endogenous α-synuclein aggregation, while others induced cell death via disruption of cellular ion homeostasis.Citation72,Citation140
Another study has confirmed that the mechanism of internalization and propagation may depend on the assembly state of the protein: oligomers and fibrils were uptaken into cells via endocytosis, while the monomeric protein passively crossed the plasma membrane.Citation138 Internalization of fibrillar α-synuclein required physiological temperatures and dynamin-1 activity, which were necessary for endocytosis. Moreover, it has been suggested that extracellular fibrils were internalized by receptor-mediated endocytosis. Internalized aggregates moved through the endosomal pathway and were degraded by lysosomes.Citation138 Internalization of α-synuclein preformed fibrils was increased by the presence of wheat germ agglutinin, which bound N-acetyl-glucosamine and sialic acids at the cell surface and induced adsorptive-mediated endocytosis, suggesting that some glycoproteins might be important players in fibril internalization.Citation131
Once α-synuclein aggregates reach acceptor cells, they could seed aggregation of endogenous protein in a prion-like fashion and eventually the newly formed aggregates may be released into the extracellular space. Vesicle-mediated exocytosis could be responsible for release of α-synuclein aggregates. Multiple forms of α-synuclein have been detected in the cerebrospinal fluid, blood plasma and saliva, suggesting an underlying secretory process.Citation141 It has been shown that cultured neuronal cells secreted α-synuclein monomers and aggregates by non-classical vesicle-mediated exocytosis. Moreover, the secretion was elevated in response to proteasomal and mitochondrial dysfunctions associated with PD pathogenesis.Citation142
Recently, it has been reported that fibrillar α-synuclein was transferred along axons through anterograde axonal transport, released, and subsequently uptaken by additional neurons. The axonal transfer of fibrils may explain the spread of LB between anatomically connected brain areas.Citation143
Moreover, tunneling nanotubes (TNTs) have been suggested to play a major role in the propagation of α-synuclein pathology. TNTs are tubular membrane bridges interconnecting cells over long distances, thus enabling the exchange of molecules and cytoplasmic content between cells.Citation144 TNTs could be involved in the intracellular propagation of PrPSc.Citation145 Till now, TNTs have not been implicated in PD but their involvement in the spread of α-synuclein pathology cannot be ruled out.
Toxicity of α-Synuclein Aggregates
An increasing number of studies, both in vitro and in vivo, have suggested that prefibrillar oligomers and protofibrils, rather than mature fibrils, may feature toxic species of α-synuclein and may cause neuronal injury and cell death.Citation146-Citation149 The process of fibril formation, rather than the fibrils themselves, has been suggested as a key element in α-synuclein toxicity. The presence of some α-synuclein prefibrillar species may indicate an attempt by neurons to convert toxic oligomers to fibrils, which are more stable, less dynamic structures and exhibit reduced toxicity.Citation147
Membrane permeabilization by oligomeric species represents a potential mechanism of α-synuclein mediated cytotoxicity. Monomeric α-synuclein was found to bind negatively charged phospholipid membranes and undergo the structural transition into α-helix.Citation150,Citation151 It has been shown that the protofibrillar α-synuclein species binds negatively charged vesicles more strongly than the monomer. Moreover, protofibrillar species, but not monomeric protein or fibrils, have permeabilized negatively charged vesicles to calcium.Citation150 Two PD-linked variants, A53T and A30P, show greater permeabilizing activity than WT protein, while this activity is reduced for E46K.Citation118,Citation124
The mechanism of membrane permeabilization is not clear, but protofibrillar oligomers have been suggested to permeabilize membranes in a pore-like fashion.Citation124 They may integrate into the membrane, creating pores or channel-like structures causing uncontrolled membrane permeability. Alternatively, oligomers or protofibrils could increase the conductivity of the membrane without the formation of pores.Citation152
Membrane permeabilization may initiate several pathological events leading to changed ion homeostasis, induction of oxidative stress, altered signaling pathways, dysfunction of mitochondria, inflammation and eventually cell death.Citation153 In fact, several works have found that exogenous oligomers led to an increase in intracellular Ca2+ level, diminished levels of some synaptic proteins, production of reactive oxygen species and cell death.Citation131,Citation149,Citation150,Citation154 Moreover, in mice intracerebro-ventricular injections of α-synuclein oligomers increased the activity of calcineurin, a key negative regulator of synaptic plasticity and memory function, thus resulting in a decrease of synaptic plasticity and memory deficits.Citation149
It seems that toxicity of α-synuclein oligomers may depend on their structure. Danzer and collaborators developed novel methods for generating different α-synuclein oligomeric species, which have been tested for their biological effects on SH-SY5Y cells.Citation72 Only spherical oligomers, 2 to 6 nm in height, increased intracellular calcium level and led to caspase-3 activation, suggesting that some structural properties of oligomeric α-synuclein may be important to induce toxicity.Citation72 Moreover, Bousset and collaborators have tested the toxicity of two α-synuclein assemblies (fibrils and ribbons) with different structural properties:Citation65 fibrils resulted more toxic than ribbons. In fact, fibrils induced activation of caspase-3, production of ROS and permeabilized lipid vesicles to a higher extent than ribbons.Citation65
Although large sets of data suggest that prefibrillar assemblies represent toxic species of α-synuclein, it has been shown that a homogenous population of fibrils, rather than their precursor on-assembly pathway oligomers, is highly toxic to cells.Citation155 Fibrils have been shown to permeabilize membrane vesicles and to alter calcium homeostasis. Moreover, cells exposure to increasing concentrations of fibrils resulted in the activation of caspase-3 in a concentration-dependent manner and cell death, whereas oligomeric α-synuclein did not affect cell viability and did not activate caspase-3. The authors argued that the discrepancy with previous results on oligomer toxicity originates from the comparison of protein particles at different concentrations. In previous reports, the concentration of different aggregates was usually expressed as a function of the initial soluble protein concentration and not as a real concentration of particles in solution.Citation155
Conclusions
Increasing evidence indicates that aggregation of α-synuclein is a critical factor in the etiology of PD and other synucleinopathies. Several studies have focused on explaining the structural features of α-synuclein and the mechanism leading to its aggregation and fibrillization. These investigations may explain molecular mechanisms occurring in living cells and leading to disease development. In vitro aggregation assays are powerful tools for studying the mechanisms responsible for the formation of different aggregate species of α-synuclein, their structural properties and biological functions. Moreover, aggregation assays are crucial to develop new drug compounds or novel therapy strategies to prevent disease development and progression. Recent studies clearly indicate that misfolded α-synuclein aggregates may be internalized by cells and propagated in a prion-like manner, leading to toxicity. In fact, Watts and collaborators have shown that α-synuclein aggregates formed in the brains of MSA patients are transmissible in a prion-like fashion.Citation156
Prion-like transmission and propagation of misfolded proteins seems to be a process common to several neurodegenerative disorders, not only PD, but also Alzheimer`s disease, Huntington`s disease, amyloid lateral sclerosis, and others.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
This work was supported by the European Union’s Seventh Framework Programme (FP7/2007–2013) under grant agreement n° 222887—the PRIORITY project.
Related Research Data
References
- Zhang L, Zhang C, Zhu Y, Cai Q, Chan P, Uéda K, Yu S, Yang H. Semi-quantitative analysis of alpha-synuclein in subcellular pools of rat brain neurons: an immunogold electron microscopic study using a C-terminal specific monoclonal antibody. Brain Res 2008; 1244:40 - 52; http://dx.doi.org/10.1016/j.brainres.2008.08.067; PMID: 18817762
- Maroteaux L, Scheller RH. The rat brain synucleins; family of proteins transiently associated with neuronal membrane. Brain Res Mol Brain Res 1991; 11:335 - 43; http://dx.doi.org/10.1016/0169-328X(91)90043-W; PMID: 1661825
- Iwai A, Masliah E, Yoshimoto M, Ge N, Flanagan L, de Silva HA, Kittel A, Saitoh T. The precursor protein of non-A beta component of Alzheimer’s disease amyloid is a presynaptic protein of the central nervous system. Neuron 1995; 14:467 - 75; http://dx.doi.org/10.1016/0896-6273(95)90302-X; PMID: 7857654
- Maroteaux L, Campanelli JT, Scheller RH. Synuclein: a neuron-specific protein localized to the nucleus and presynaptic nerve terminal. J Neurosci 1988; 8:2804 - 15; PMID: 3411354
- Mori F, Tanji K, Yoshimoto M, Takahashi H, Wakabayashi K. Demonstration of alpha-synuclein immunoreactivity in neuronal and glial cytoplasm in normal human brain tissue using proteinase K and formic acid pretreatment. Exp Neurol 2002; 176:98 - 104; http://dx.doi.org/10.1006/exnr.2002.7929; PMID: 12093086
- Papp MI, Komoly S. Filamentous Glial Cytoplasmic Inclusions in the Cns of Patients with Various Combinations of Striatonigral Degeneration (Snd), Olivopontocerebellar Atrophy (Opca) and Shy-Drager Syndrome (Sds). Clin Neuropathol 1988; 7:195
- Lee HJ, Lee SJ. Characterization of cytoplasmic alpha-synuclein aggregates. Fibril formation is tightly linked to the inclusion-forming process in cells. J Biol Chem 2002; 277:48976 - 83; http://dx.doi.org/10.1074/jbc.M208192200; PMID: 12351642
- Baptista MJ, O’Farrell C, Daya S, Ahmad R, Miller DW, Hardy J, Farrer MJ, Cookson MR. Co-ordinate transcriptional regulation of dopamine synthesis genes by alpha-synuclein in human neuroblastoma cell lines. J Neurochem 2003; 85:957 - 68; http://dx.doi.org/10.1046/j.1471-4159.2003.01742.x; PMID: 12716427
- Yavich L, Tanila H, Vepsäläinen S, Jäkälä P. Role of alpha-synuclein in presynaptic dopamine recruitment. J Neurosci 2004; 24:11165 - 70; http://dx.doi.org/10.1523/JNEUROSCI.2559-04.2004; PMID: 15590933
- Fountaine TM, Wade-Martins R. RNA interference-mediated knockdown of alpha-synuclein protects human dopaminergic neuroblastoma cells from MPP(+) toxicity and reduces dopamine transport. J Neurosci Res 2007; 85:351 - 63; http://dx.doi.org/10.1002/jnr.21125; PMID: 17131421
- Lee FJ, Liu F, Pristupa ZB, Niznik HB. Direct binding and functional coupling of alpha-synuclein to the dopamine transporters accelerate dopamine-induced apoptosis. FASEB J 2001; 15:916 - 26; http://dx.doi.org/10.1096/fj.00-0334com; PMID: 11292651
- Wersinger C, Prou D, Vernier P, Sidhu A. Modulation of dopamine transporter function by alpha-synuclein is altered by impairment of cell adhesion and by induction of oxidative stress. FASEB J 2003; 17:2151 - 3; PMID: 12958153
- Wersinger C, Sidhu A. Attenuation of dopamine transporter activity by alpha-synuclein. Neurosci Lett 2003; 340:189 - 92; http://dx.doi.org/10.1016/S0304-3940(03)00097-1; PMID: 12672538
- Sidhu A, Wersinger C, Moussa CE, Vernier P. The role of alpha-synuclein in both neuroprotection and neurodegeneration. Ann N Y Acad Sci 2004; 1035:250 - 70; http://dx.doi.org/10.1196/annals.1332.016; PMID: 15681812
- Chandra S, Gallardo G, Fernández-Chacón R, Schlüter OM, Südhof TC. Alpha-synuclein cooperates with CSPalpha in preventing neurodegeneration. Cell 2005; 123:383 - 96; http://dx.doi.org/10.1016/j.cell.2005.09.028; PMID: 16269331
- Deleersnijder A, Gerard M, Debyser Z, Baekelandt V. The remarkable conformational plasticity of alpha-synuclein: blessing or curse?. Trends Mol Med 2013; 19:368 - 77; http://dx.doi.org/10.1016/j.molmed.2013.04.002; PMID: 23648364
- Tompa P. Intrinsically unstructured proteins evolve by repeat expansion. Bioessays 2003; 25:847 - 55; http://dx.doi.org/10.1002/bies.10324; PMID: 12938174
- Tompa P. Intrinsically unstructured proteins. Trends Biochem Sci 2002; 27:527 - 33; http://dx.doi.org/10.1016/S0968-0004(02)02169-2; PMID: 12368089
- Bisaglia M, Tessari I, Pinato L, Bellanda M, Giraudo S, Fasano M, Bergantino E, Bubacco L, Mammi S. A topological model of the interaction between alpha-synuclein and sodium dodecyl sulfate micelles. Biochemistry 2005; 44:329 - 39; http://dx.doi.org/10.1021/bi048448q; PMID: 15628875
- Davidson WS, Jonas A, Clayton DF, George JM. Stabilization of alpha-synuclein secondary structure upon binding to synthetic membranes. J Biol Chem 1998; 273:9443 - 9; http://dx.doi.org/10.1074/jbc.273.16.9443; PMID: 9545270
- Eliezer D, Kutluay E, Bussell R Jr., Browne G. Conformational properties of alpha-synuclein in its free and lipid-associated states. J Mol Biol 2001; 307:1061 - 73; http://dx.doi.org/10.1006/jmbi.2001.4538; PMID: 11286556
- Chandra S, Chen X, Rizo J, Jahn R, Südhof TC. A broken alpha -helix in folded alpha -Synuclein. J Biol Chem 2003; 278:15313 - 8; http://dx.doi.org/10.1074/jbc.M213128200; PMID: 12586824
- Ulmer TS, Bax A, Cole NB, Nussbaum RL. Structure and dynamics of micelle-bound human alpha-synuclein. J Biol Chem 2005; 280:9595 - 603; http://dx.doi.org/10.1074/jbc.M411805200; PMID: 15615727
- Rao JN, Jao CC, Hegde BG, Langen R, Ulmer TS. A combinatorial NMR and EPR approach for evaluating the structural ensemble of partially folded proteins. J Am Chem Soc 2010; 132:8657 - 68; http://dx.doi.org/10.1021/ja100646t; PMID: 20524659
- Gruschus JM, Yap TL, Pistolesi S, Maltsev AS, Lee JC. NMR structure of calmodulin complexed to an N-terminally acetylated α-synuclein peptide. Biochemistry 2013; 52:3436 - 45; http://dx.doi.org/10.1021/bi400199p; PMID: 23607618
- Xie YY, Zhou CJ, Zhou ZR, Hong J, Che MX, Fu QS, Song AX, Lin DH, Hu HY. Interaction with synphilin-1 promotes inclusion formation of alpha-synuclein: mechanistic insights and pathological implication. FASEB J 2010; 24:196 - 205; http://dx.doi.org/10.1096/fj.09-133082; PMID: 19762560
- De Genst EJ, Guilliams T, Wellens J, O’Day EM, Waudby CA, Meehan S, Dumoulin M, Hsu ST, Cremades N, Verschueren KH, et al. Structure and properties of a complex of α-synuclein and a single-domain camelid antibody. J Mol Biol 2010; 402:326 - 43; http://dx.doi.org/10.1016/j.jmb.2010.07.001; PMID: 20620148
- Zhao M, Cascio D, Sawaya MR, Eisenberg D. Structures of segments of α-synuclein fused to maltose-binding protein suggest intermediate states during amyloid formation. Protein Sci 2011; 20:996 - 1004; http://dx.doi.org/10.1002/pro.630; PMID: 21462277
- Bartels T, Choi JG, Selkoe DJ. α-Synuclein occurs physiologically as a helically folded tetramer that resists aggregation. Nature 2011; 477:107 - 10; http://dx.doi.org/10.1038/nature10324; PMID: 21841800
- Fauvet B, Mbefo MK, Fares MB, Desobry C, Michael S, Ardah MT, Tsika E, Coune P, Prudent M, Lion N, et al. α-Synuclein in central nervous system and from erythrocytes, mammalian cells, and Escherichia coli exists predominantly as disordered monomer. J Biol Chem 2012; 287:15345 - 64; http://dx.doi.org/10.1074/jbc.M111.318949; PMID: 22315227
- Spillantini MG, Crowther RA, Jakes R, Hasegawa M, Goedert M. alpha-Synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and dementia with lewy bodies. Proc Natl Acad Sci U S A 1998; 95:6469 - 73; http://dx.doi.org/10.1073/pnas.95.11.6469; PMID: 9600990
- Conway KA, Harper JD, Lansbury PT. Accelerated in vitro fibril formation by a mutant alpha-synuclein linked to early-onset Parkinson disease. Nat Med 1998; 4:1318 - 20; http://dx.doi.org/10.1038/3311; PMID: 9809558
- Forno LS, Langston JW. Lewy Bodies and Aging. J Neuropathol Exp Neurol 1990; 49:278; http://dx.doi.org/10.1097/00005072-199005000-00058
- Serpell LC, Berriman J, Jakes R, Goedert M, Crowther RA. Fiber diffraction of synthetic alpha-synuclein filaments shows amyloid-like cross-beta conformation. Proc Natl Acad Sci U S A 2000; 97:4897 - 902; http://dx.doi.org/10.1073/pnas.97.9.4897; PMID: 10781096
- Weinreb PH, Zhen W, Poon AW, Conway KA, Lansbury PT Jr.. NACP, a protein implicated in Alzheimer’s disease and learning, is natively unfolded. Biochemistry 1996; 35:13709 - 15; http://dx.doi.org/10.1021/bi961799n; PMID: 8901511
- Giasson BI, Uryu K, Trojanowski JQ, Lee VM. Mutant and wild type human alpha-synucleins assemble into elongated filaments with distinct morphologies in vitro. J Biol Chem 1999; 274:7619 - 22; http://dx.doi.org/10.1074/jbc.274.12.7619; PMID: 10075647
- Narhi L, Wood SJ, Steavenson S, Jiang Y, Wu GM, Anafi D, Kaufman SA, Martin F, Sitney K, Denis P, et al. Both familial Parkinson’s disease mutations accelerate alpha-synuclein aggregation. J Biol Chem 1999; 274:9843 - 6; http://dx.doi.org/10.1074/jbc.274.14.9843; PMID: 10092675
- Ren G, Wang X, Hao S, Hu H, Wang CC. Translocation of alpha-synuclein expressed in Escherichia coli. J Bacteriol 2007; 189:2777 - 86; http://dx.doi.org/10.1128/JB.01406-06; PMID: 17277073
- Huang C, Ren G, Zhou H, Wang CC. A new method for purification of recombinant human alpha-synuclein in Escherichia coli. Protein Expr Purif 2005; 42:173 - 7; http://dx.doi.org/10.1016/j.pep.2005.02.014; PMID: 15939304
- Nilsson MR. Techniques to study amyloid fibril formation in vitro. Methods 2004; 34:151 - 60; http://dx.doi.org/10.1016/j.ymeth.2004.03.012; PMID: 15283924
- Uversky VN, Li J, Fink AL. Evidence for a partially folded intermediate in alpha-synuclein fibril formation. J Biol Chem 2001; 276:10737 - 44; http://dx.doi.org/10.1074/jbc.M010907200; PMID: 11152691
- Giehm L, Lorenzen N, Otzen DE. Assays for α-synuclein aggregation. Methods 2011; 53:295 - 305; http://dx.doi.org/10.1016/j.ymeth.2010.12.008; PMID: 21163351
- Eisert R, Felau L, Brown LR. Methods for enhancing the accuracy and reproducibility of Congo red and thioflavin T assays. Anal Biochem 2006; 353:144 - 6; http://dx.doi.org/10.1016/j.ab.2006.03.015; PMID: 16620754
- Masuda M, Suzuki N, Taniguchi S, Oikawa T, Nonaka T, Iwatsubo T, Hisanaga S, Goedert M, Hasegawa M. Small molecule inhibitors of alpha-synuclein filament assembly. Biochemistry 2006; 45:6085 - 94; http://dx.doi.org/10.1021/bi0600749; PMID: 16681381
- Frid P, Anisimov SV, Popovic N. Congo red and protein aggregation in neurodegenerative diseases. Brain Res Rev 2007; 53:135 - 60; http://dx.doi.org/10.1016/j.brainresrev.2006.08.001; PMID: 16959325
- Celej MS, Jares-Erijman EA, Jovin TM. Fluorescent N-arylaminonaphthalene sulfonate probes for amyloid aggregation of alpha-synuclein. Biophys J 2008; 94:4867 - 79; http://dx.doi.org/10.1529/biophysj.107.125211; PMID: 18339734
- Del Mercato LL, Pompa PP, Maruccio G, Della Torre A, Sabella S, Tamburro AM, Cingolani R, Rinaldi R. Charge transport and intrinsic fluorescence in amyloid-like fibrils. Proc Natl Acad Sci U S A 2007; 104:18019 - 24; http://dx.doi.org/10.1073/pnas.0702843104; PMID: 17984067
- Chan FTS, Kaminski Schierle GS, Kumita JR, Bertoncini CW, Dobson CM, Kaminski CF. Protein amyloids develop an intrinsic fluorescence signature during aggregation. Analyst 2013; 138:2156 - 62; http://dx.doi.org/10.1039/c3an36798c; PMID: 23420088
- Sharpe S, Simonetti K, Yau J, Walsh P. Solid-State NMR characterization of autofluorescent fibrils formed by the elastin-derived peptide GVGVAGVG. Biomacromolecules 2011; 12:1546 - 55; http://dx.doi.org/10.1021/bm101486s; PMID: 21456595
- Pinotsi D, Buell AK, Dobson CM, Kaminski Schierle GS, Kaminski CF. A label-free, quantitative assay of amyloid fibril growth based on intrinsic fluorescence. Chembiochem 2013; 14:846 - 50; http://dx.doi.org/10.1002/cbic.201300103; PMID: 23592254
- Rochet JC, Lansbury PT Jr.. Amyloid fibrillogenesis: themes and variations. Curr Opin Struct Biol 2000; 10:60 - 8; http://dx.doi.org/10.1016/S0959-440X(99)00049-4; PMID: 10679462
- Uversky VN, Gillespie JR, Fink AL. Why are “natively unfolded” proteins unstructured under physiologic conditions?. Proteins 2000; 41:415 - 27; http://dx.doi.org/10.1002/1097-0134(20001115)41:3<415::AID-PROT130>3.0.CO;2-7; PMID: 11025552
- Baldwin RL. Temperature dependence of the hydrophobic interaction in protein folding. Proc Natl Acad Sci U S A 1986; 83:8069 - 72; http://dx.doi.org/10.1073/pnas.83.21.8069; PMID: 3464944
- Wood SJ, Wypych J, Steavenson S, Louis JC, Citron M, Biere AL. alpha-synuclein fibrillogenesis is nucleation-dependent. Implications for the pathogenesis of Parkinson’s disease. J Biol Chem 1999; 274:19509 - 12; http://dx.doi.org/10.1074/jbc.274.28.19509; PMID: 10391881
- Conway KA, Lee SJ, Rochet JC, Ding TT, Williamson RE, Lansbury PT Jr.. Acceleration of oligomerization, not fibrillization, is a shared property of both alpha-synuclein mutations linked to early-onset Parkinson’s disease: implications for pathogenesis and therapy. Proc Natl Acad Sci U S A 2000; 97:571 - 6; http://dx.doi.org/10.1073/pnas.97.2.571; PMID: 10639120
- Nonaka T, Watanabe ST, Iwatsubo T, Hasegawa M. Seeded aggregation and toxicity of alpha-synuclein and tau: cellular models of neurodegenerative diseases. J Biol Chem 2010; 285:34885 - 98; http://dx.doi.org/10.1074/jbc.M110.148460; PMID: 20805224
- Nielsen L, Khurana R, Coats A, Frokjaer S, Brange J, Vyas S, Uversky VN, Fink AL. Effect of environmental factors on the kinetics of insulin fibril formation: elucidation of the molecular mechanism. Biochemistry 2001; 40:6036 - 46; http://dx.doi.org/10.1021/bi002555c; PMID: 11352739
- Giehm L, Otzen DE. Strategies to increase the reproducibility of protein fibrillization in plate reader assays. Anal Biochem 2010; 400:270 - 81; http://dx.doi.org/10.1016/j.ab.2010.02.001; PMID: 20149780
- Horvath I, Weise CF, Andersson EK, Chorell E, Sellstedt M, Bengtsson C, Olofsson A, Hultgren SJ, Chapman M, Wolf-Watz M, et al. Mechanisms of protein oligomerization: inhibitor of functional amyloids templates α-synuclein fibrillation. J Am Chem Soc 2012; 134:3439 - 44; http://dx.doi.org/10.1021/ja209829m; PMID: 22260746
- Conway KA, Harper JD, Lansbury PT Jr.. Fibrils formed in vitro from alpha-synuclein and two mutant forms linked to Parkinson’s disease are typical amyloid. Biochemistry 2000; 39:2552 - 63; http://dx.doi.org/10.1021/bi991447r; PMID: 10704204
- Tycko R. Solid-state NMR studies of amyloid fibril structure. Annu Rev Phys Chem 2011; 62:279 - 99; http://dx.doi.org/10.1146/annurev-physchem-032210-103539; PMID: 21219138
- Chen M, Margittai M, Chen J, Langen R. Investigation of alpha-synuclein fibril structure by site-directed spin labeling. J Biol Chem 2007; 282:24970 - 9; http://dx.doi.org/10.1074/jbc.M700368200; PMID: 17573347
- Pornsuwan S, Giller K, Riedel D, Becker S, Griesinger C, Bennati M. Long-range distances in amyloid fibrils of α-synuclein from PELDOR spectroscopy. Angew Chem Int Ed Engl 2013; 52:10290 - 4; http://dx.doi.org/10.1002/anie.201304747; PMID: 23934970
- Lv G, Kumar A, Giller K, Orcellet ML, Riedel D, Fernández CO, Becker S, Lange A. Structural comparison of mouse and human α-synuclein amyloid fibrils by solid-state NMR. J Mol Biol 2012; 420:99 - 111; http://dx.doi.org/10.1016/j.jmb.2012.04.009; PMID: 22516611
- Bousset L, Pieri L, Ruiz-Arlandis G, Gath J, Jensen PH, Habenstein B, Madiona K, Olieric V, Böckmann A, Meier BH, et al. Structural and functional characterization of two alpha-synuclein strains. Nat Commun 2013; 4:2575; http://dx.doi.org/10.1038/ncomms3575; PMID: 24108358
- Kloepper KD, Woods WS, Winter KA, George JM, Rienstra CM. Preparation of alpha-synuclein fibrils for solid-state NMR: expression, purification, and incubation of wild-type and mutant forms. Protein Expr Purif 2006; 48:112 - 7; http://dx.doi.org/10.1016/j.pep.2006.02.009; PMID: 16564705
- Heise H, Celej MS, Becker S, Riedel D, Pelah A, Kumar A, Jovin TM, Baldus M. Solid-state NMR reveals structural differences between fibrils of wild-type and disease-related A53T mutant alpha-synuclein. J Mol Biol 2008; 380:444 - 50; http://dx.doi.org/10.1016/j.jmb.2008.05.026; PMID: 18539297
- Heise H, Hoyer W, Becker S, Andronesi OC, Riedel D, Baldus M. Molecular-level secondary structure, polymorphism, and dynamics of full-length alpha-synuclein fibrils studied by solid-state NMR. Proc Natl Acad Sci U S A 2005; 102:15871 - 6; http://dx.doi.org/10.1073/pnas.0506109102; PMID: 16247008
- Vilar M, Chou HT, Lührs T, Maji SK, Riek-Loher D, Verel R, Manning G, Stahlberg H, Riek R. The fold of alpha-synuclein fibrils. Proc Natl Acad Sci U S A 2008; 105:8637 - 42; http://dx.doi.org/10.1073/pnas.0712179105; PMID: 18550842
- Comellas G, Lemkau LR, Nieuwkoop AJ, Kloepper KD, Ladror DT, Ebisu R, Woods WS, Lipton AS, George JM, Rienstra CM. Structured regions of α-synuclein fibrils include the early-onset Parkinson’s disease mutation sites. J Mol Biol 2011; 411:881 - 95; http://dx.doi.org/10.1016/j.jmb.2011.06.026; PMID: 21718702
- Gath J, Habenstein B, Bousset L, Melki R, Meier BH, Böckmann A. Solid-state NMR sequential assignments of α-synuclein. Biomol NMR Assign 2012; 6:51 - 5; http://dx.doi.org/10.1007/s12104-011-9324-3; PMID: 21744165
- Danzer KM, Haasen D, Karow AR, Moussaud S, Habeck M, Giese A, Kretzschmar H, Hengerer B, Kostka M. Different species of alpha-synuclein oligomers induce calcium influx and seeding. J Neurosci 2007; 27:9220 - 32; http://dx.doi.org/10.1523/JNEUROSCI.2617-07.2007; PMID: 17715357
- Munishkina LA, Phelan C, Uversky VN, Fink AL. Conformational behavior and aggregation of alpha-synuclein in organic solvents: modeling the effects of membranes. Biochemistry 2003; 42:2720 - 30; http://dx.doi.org/10.1021/bi027166s; PMID: 12614167
- Munishkina LA, Cooper EM, Uversky VN, Fink AL. The effect of macromolecular crowding on protein aggregation and amyloid fibril formation. J Mol Recognit 2004; 17:456 - 64; http://dx.doi.org/10.1002/jmr.699; PMID: 15362105
- Li J, Uversky VN, Fink AL. Effect of familial Parkinson’s disease point mutations A30P and A53T on the structural properties, aggregation, and fibrillation of human alpha-synuclein. Biochemistry 2001; 40:11604 - 13; http://dx.doi.org/10.1021/bi010616g; PMID: 11560511
- Uversky VN, Li J, Fink AL. Metal-triggered structural transformations, aggregation, and fibrillation of human alpha-synuclein. A possible molecular NK between Parkinson’s disease and heavy metal exposure. J Biol Chem 2001; 276:44284 - 96; http://dx.doi.org/10.1074/jbc.M105343200; PMID: 11553618
- Goers J, Uversky VN, Fink AL. Polycation-induced oligomerization and accelerated fibrillation of human alpha-synuclein in vitro. Protein Sci 2003; 12:702 - 7; http://dx.doi.org/10.1110/ps.0230903; PMID: 12649428
- Luk KC, Song C, O’Brien P, Stieber A, Branch JR, Brunden KR, Trojanowski JQ, Lee VM. Exogenous alpha-synuclein fibrils seed the formation of Lewy body-like intracellular inclusions in cultured cells. Proc Natl Acad Sci U S A 2009; 106:20051 - 6; http://dx.doi.org/10.1073/pnas.0908005106; PMID: 19892735
- Masuda-Suzukake M, Nonaka T, Hosokawa M, Oikawa T, Arai T, Akiyama H, Mann DM, Hasegawa M. Prion-like spreading of pathological α-synuclein in brain. Brain 2013; 136:1128 - 38; http://dx.doi.org/10.1093/brain/awt037; PMID: 23466394
- van Raaij ME, van Gestel J, Segers-Nolten IMJ, de Leeuw SW, Subramaniam V. Concentration dependence of alpha-synuclein fibril length assessed by quantitative atomic force microscopy and statistical-mechanical theory. Biophys J 2008; 95:4871 - 8; http://dx.doi.org/10.1529/biophysj.107.127464; PMID: 18676659
- Powers ET, Powers DL. The kinetics of nucleated polymerizations at high concentrations: amyloid fibril formation near and above the “supercritical concentration”. Biophys J 2006; 91:122 - 32; http://dx.doi.org/10.1529/biophysj.105.073767; PMID: 16603497
- Fink AL. Factors Affecting the Fibrillation of alpha-synuclein, a natively unfolded protein. In Misbehaving Proteins: Protein (Mis)Folding, Aggregation, and Stability. Springer, 2006.
- Shtilerman MD, Ding TT, Lansbury PT Jr.. Molecular crowding accelerates fibrillization of alpha-synuclein: could an increase in the cytoplasmic protein concentration induce Parkinson’s disease?. Biochemistry 2002; 41:3855 - 60; http://dx.doi.org/10.1021/bi0120906; PMID: 11900526
- Fink AL. Protein aggregation: folding aggregates, inclusion bodies and amyloid. Fold Des 1998; 3:R9 - 23; http://dx.doi.org/10.1016/S1359-0278(98)00002-9; PMID: 9502314
- Uversky VN, Li J, Fink AL. Pesticides directly accelerate the rate of alpha-synuclein fibril formation: a possible factor in Parkinson’s disease. FEBS Lett 2001; 500:105 - 8; http://dx.doi.org/10.1016/S0014-5793(01)02597-2; PMID: 11445065
- Serio TR, Cashikar AG, Kowal AS, Sawicki GJ, Moslehi JJ, Serpell L, Arnsdorf MF, Lindquist SL. Nucleated conformational conversion and the replication of conformational information by a prion determinant. Science 2000; 289:1317 - 21; http://dx.doi.org/10.1126/science.289.5483.1317; PMID: 10958771
- Cohlberg JA, Li J, Uversky VN, Fink AL. Heparin and other glycosaminoglycans stimulate the formation of amyloid fibrils from alpha-synuclein in vitro. Biochemistry 2002; 41:1502 - 11; http://dx.doi.org/10.1021/bi011711s; PMID: 11814343
- Zhu M, Rajamani S, Kaylor J, Han S, Zhou F, Fink AL. The flavonoid baicalein inhibits fibrillation of alpha-synuclein and disaggregates existing fibrils. J Biol Chem 2004; 279:26846 - 57; http://dx.doi.org/10.1074/jbc.M403129200; PMID: 15096521
- Manning-Bog AB, McCormack AL, Li J, Uversky VN, Fink AL, Di Monte DA. The herbicide paraquat causes up-regulation and aggregation of alpha-synuclein in mice: paraquat and alpha-synuclein. J Biol Chem 2002; 277:1641 - 4; http://dx.doi.org/10.1074/jbc.C100560200; PMID: 11707429
- Olanow CW, Tatton WG. Etiology and pathogenesis of Parkinson’s disease. Annu Rev Neurosci 1999; 22:123 - 44; http://dx.doi.org/10.1146/annurev.neuro.22.1.123; PMID: 10202534
- Uversky VN. Neuropathology, biochemistry, and biophysics of alpha-synuclein aggregation. J Neurochem 2007; 103:17 - 37; PMID: 17623039
- Uversky VN, Eliezer D. Biophysics of Parkinson’s disease: structure and aggregation of alpha-synuclein. Curr Protein Pept Sci 2009; 10:483 - 99; http://dx.doi.org/10.2174/138920309789351921; PMID: 19538146
- Betarbet R, Sherer TB, MacKenzie G, Garcia-Osuna M, Panov AV, Greenamyre JT. Chronic systemic pesticide exposure reproduces features of Parkinson’s disease. Nat Neurosci 2000; 3:1301 - 6; http://dx.doi.org/10.1038/81834; PMID: 11100151
- Giasson BI, Lee VM. A new link between pesticides and Parkinson’s disease. Nat Neurosci 2000; 3:1227 - 8; http://dx.doi.org/10.1038/81737; PMID: 11100135
- Natalello A, Benetti F, Doglia SM, Legname G, Grandori R. Compact conformations of α-synuclein induced by alcohols and copper. Proteins 2011; 79:611 - 21; http://dx.doi.org/10.1002/prot.22909; PMID: 21120859
- Zayed J, Ducic S, Campanella G, Panisset JC, André P, Masson H, Roy M. [Environmental factors in the etiology of Parkinson’s disease]. Can J Neurol Sci 1990; 17:286 - 91; PMID: 2207882
- Rybicki BA, Johnson CC, Uman J, Gorell JM. Parkinson’s disease mortality and the industrial use of heavy metals in Michigan. Mov Disord 1993; 8:87 - 92; http://dx.doi.org/10.1002/mds.870080116; PMID: 8419812
- Gorell JM, Johnson CC, Rybicki BA, Peterson EL, Kortsha GX, Brown GG, Richardson RJ. Occupational exposures to metals as risk factors for Parkinson’s disease. Neurology 1997; 48:650 - 8; http://dx.doi.org/10.1212/WNL.48.3.650; PMID: 9065542
- Gorell JM, Johnson CC, Rybicki BA, Peterson EL, Kortsha GX, Brown GG, Richardson RJ. Occupational exposure to manganese, copper, lead, iron, mercury and zinc and the risk of Parkinson’s disease. Neurotoxicology 1999; 20:239 - 47; PMID: 10385887
- Hirsch EC, Brandel JP, Galle P, Javoy-Agid F, Agid Y. Iron and aluminum increase in the substantia nigra of patients with Parkinson’s disease: an X-ray microanalysis. J Neurochem 1991; 56:446 - 51; http://dx.doi.org/10.1111/j.1471-4159.1991.tb08170.x; PMID: 1988548
- Dexter DT, Carayon A, Javoy-Agid F, Agid Y, Wells FR, Daniel SE, Lees AJ, Jenner P, Marsden CD. Alterations in the levels of iron, ferritin and other trace metals in Parkinson’s disease and other neurodegenerative diseases affecting the basal ganglia. Brain 1991; 114:1953 - 75; http://dx.doi.org/10.1093/brain/114.4.1953; PMID: 1832073
- Dexter DT, Wells FR, Lees AJ, Agid F, Agid Y, Jenner P, Marsden CD. Increased nigral iron content and alterations in other metal ions occurring in brain in Parkinson’s disease. J Neurochem 1989; 52:1830 - 6; http://dx.doi.org/10.1111/j.1471-4159.1989.tb07264.x; PMID: 2723638
- Goers J, Manning-Bog AB, McCormack AL, Millett IS, Doniach S, Di Monte DA, Uversky VN, Fink AL. Nuclear localization of alpha-synuclein and its interaction with histones. Biochemistry 2003; 42:8465 - 71; http://dx.doi.org/10.1021/bi0341152; PMID: 12859192
- Meng X, Munishkina LA, Fink AL, Uversky VN. Effects of Various Flavonoids on the α-Synuclein Fibrillation Process. Parkinsons Dis 2010; 2010:650794; http://dx.doi.org/10.4061/2010/650794; PMID: 20976092
- Rekas A, Adda CG, Andrew Aquilina J, Barnham KJ, Sunde M, Galatis D, Williamson NA, Masters CL, Anders RF, Robinson CV, et al. Interaction of the molecular chaperone alphaB-crystallin with alpha-synuclein: effects on amyloid fibril formation and chaperone activity. J Mol Biol 2004; 340:1167 - 83; http://dx.doi.org/10.1016/j.jmb.2004.05.054; PMID: 15236975
- Dedmon MM, Christodoulou J, Wilson MR, Dobson CM. Heat shock protein 70 inhibits alpha-synuclein fibril formation via preferential binding to prefibrillar species. J Biol Chem 2005; 280:14733 - 40; http://dx.doi.org/10.1074/jbc.M413024200; PMID: 15671022
- Huang C, Cheng H, Hao S, Zhou H, Zhang X, Gao J, Sun QH, Hu H, Wang CC. Heat shock protein 70 inhibits alpha-synuclein fibril formation via interactions with diverse intermediates. J Mol Biol 2006; 364:323 - 36; http://dx.doi.org/10.1016/j.jmb.2006.08.062; PMID: 17010992
- Bruinsma IB, Bruggink KA, Kinast K, Versleijen AAM, Segers-Nolten IMJ, Subramaniam V, Kuiperij HB, Boelens W, de Waal RM, Verbeek MM. Inhibition of α-synuclein aggregation by small heat shock proteins. Proteins 2011; 79:2956 - 67; http://dx.doi.org/10.1002/prot.23152; PMID: 21905118
- Rekas A, Lo V, Gadd GE, Cappai R, Yun SI. PAMAM dendrimers as potential agents against fibrillation of alpha-synuclein, a Parkinson’s disease-related protein. Macromol Biosci 2009; 9:230 - 8; http://dx.doi.org/10.1002/mabi.200800242; PMID: 19116892
- Janeczek P, Mackay RK, Lea RA, Dodd PR, Lewohl JM. Reduced expression of α-synuclein in alcoholic brain: influence of SNCA-Rep1 genotype. Addict Biol 2012; forthcoming http://dx.doi.org/10.1111/j.1369-1600.2012.00495.x; PMID: 22974310
- Janeczek P, Lewohl JM. The role of α-synuclein in the pathophysiology of alcoholism. Neurochem Int 2013; 63:154 - 62; http://dx.doi.org/10.1016/j.neuint.2013.06.007; PMID: 23791711
- Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 1997; 276:2045 - 7; http://dx.doi.org/10.1126/science.276.5321.2045; PMID: 9197268
- Krüger R, Kuhn W, Müller T, Woitalla D, Graeber M, Kösel S, Przuntek H, Epplen JT, Schöls L, Riess O. Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson’s disease. Nat Genet 1998; 18:106 - 8; http://dx.doi.org/10.1038/ng0298-106; PMID: 9462735
- Zarranz JJ, Alegre J, Gómez-Esteban JC, Lezcano E, Ros R, Ampuero I, Vidal L, Hoenicka J, Rodriguez O, Atarés B, et al. The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann Neurol 2004; 55:164 - 73; http://dx.doi.org/10.1002/ana.10795; PMID: 14755719
- Appel-Cresswell S, Vilarino-Guell C, Encarnacion M, Sherman H, Yu I, Shah B, Weir D, Thompson C, Szu-Tu C, Trinh J, et al. Alpha-synuclein p.H50Q, a novel pathogenic mutation for Parkinson’s disease. Mov Disord 2013; 28:811 - 3; http://dx.doi.org/10.1002/mds.25421; PMID: 23457019
- Proukakis C, Dudzik CG, Brier T, MacKay DS, Cooper JM, Millhauser GL, Houlden H, Schapira AH. A novel α-synuclein missense mutation in Parkinson disease. Neurology 2013; 80:1062 - 4; http://dx.doi.org/10.1212/WNL.0b013e31828727ba; PMID: 23427326
- Greenbaum EA, Graves CL, Mishizen-Eberz AJ, Lupoli MA, Lynch DR, Englander SW, Axelsen PH, Giasson BI. The E46K mutation in alpha-synuclein increases amyloid fibril formation. J Biol Chem 2005; 280:7800 - 7; http://dx.doi.org/10.1074/jbc.M411638200; PMID: 15632170
- Fredenburg RA, Rospigliosi C, Meray RK, Kessler JC, Lashuel HA, Eliezer D, Lansbury PT Jr.. The impact of the E46K mutation on the properties of alpha-synuclein in its monomeric and oligomeric states. Biochemistry 2007; 46:7107 - 18; http://dx.doi.org/10.1021/bi7000246; PMID: 17530780
- Lemkau LR, Comellas G, Kloepper KD, Woods WS, George JM, Rienstra CM. Mutant protein A30P α-synuclein adopts wild-type fibril structure, despite slower fibrillation kinetics. J Biol Chem 2012; 287:11526 - 32; http://dx.doi.org/10.1074/jbc.M111.306902; PMID: 22334684
- Ghosh D, Mondal M, Mohite GM, Singh PK, Ranjan P, Anoop A, Ghosh S, Jha NN, Kumar A, Maji SK. The Parkinson’s disease-associated H50Q mutation accelerates α-Synuclein aggregation in vitro. Biochemistry 2013; 52:6925 - 7; http://dx.doi.org/10.1021/bi400999d; PMID: 24047453
- Choi W, Zibaee S, Jakes R, Serpell LC, Davletov B, Crowther RA, Goedert M. Mutation E46K increases phospholipid binding and assembly into filaments of human alpha-synuclein. FEBS Lett 2004; 576:363 - 8; http://dx.doi.org/10.1016/j.febslet.2004.09.038; PMID: 15498564
- Sacino AN, Thomas MA, Ceballos-Diaz C, Cruz PE, Rosario AM, Lewis J, Giasson BI, Golde TE. Conformational templating of α-synuclein aggregates in neuronal-glial cultures. Mol Neurodegener 2013; 8:17; http://dx.doi.org/10.1186/1750-1326-8-17; PMID: 23714769
- Lemkau LR, Comellas G, Lee SW, Rikardsen LK, Woods WS, George JM, Rienstra CM. Site-specific perturbations of alpha-synuclein fibril structure by the Parkinson’s disease associated mutations A53T and E46K. PLoS One 2013; 8:e49750; http://dx.doi.org/10.1371/journal.pone.0049750; PMID: 23505409
- Volles MJ, Lansbury PT Jr.. Vesicle permeabilization by protofibrillar alpha-synuclein is sensitive to Parkinson’s disease-linked mutations and occurs by a pore-like mechanism. Biochemistry 2002; 41:4595 - 602; http://dx.doi.org/10.1021/bi0121353; PMID: 11926821
- Yonetani M, Nonaka T, Masuda M, Inukai Y, Oikawa T, Hisanaga S, Hasegawa M. Conversion of wild-type alpha-synuclein into mutant-type fibrils and its propagation in the presence of A30P mutant. J Biol Chem 2009; 284:7940 - 50; http://dx.doi.org/10.1074/jbc.M807482200; PMID: 19164293
- Braak H, Del Tredici K, Rüb U, de Vos RA, Jansen Steur EN, Braak E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging 2003; 24:197 - 211; http://dx.doi.org/10.1016/S0197-4580(02)00065-9; PMID: 12498954
- Kordower JH, Chu Y, Hauser RA, Freeman TB, Olanow CW. Lewy body-like pathology in long-term embryonic nigral transplants in Parkinson’s disease. Nat Med 2008; 14:504 - 6; http://dx.doi.org/10.1038/nm1747; PMID: 18391962
- Li JY, Englund E, Holton JL, Soulet D, Hagell P, Lees AJ, Lashley T, Quinn NP, Rehncrona S, Björklund A, et al. Lewy bodies in grafted neurons in subjects with Parkinson’s disease suggest host-to-graft disease propagation. Nat Med 2008; 14:501 - 3; http://dx.doi.org/10.1038/nm1746; PMID: 18391963
- Desplats P, Lee HJ, Bae EJ, Patrick C, Rockenstein E, Crews L, Spencer B, Masliah E, Lee SJ. Inclusion formation and neuronal cell death through neuron-to-neuron transmission of alpha-synuclein. Proc Natl Acad Sci U S A 2009; 106:13010 - 5; http://dx.doi.org/10.1073/pnas.0903691106; PMID: 19651612
- Luk KC, Kehm V, Carroll J, Zhang B, O’Brien P, Trojanowski JQ, Lee VM. Pathological α-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science 2012; 338:949 - 53; http://dx.doi.org/10.1126/science.1227157; PMID: 23161999
- Volpicelli-Daley LA, Luk KC, Patel TP, Tanik SA, Riddle DM, Stieber A, Meaney DF, Trojanowski JQ, Lee VM. Exogenous α-synuclein fibrils induce Lewy body pathology leading to synaptic dysfunction and neuron death. Neuron 2011; 72:57 - 71; http://dx.doi.org/10.1016/j.neuron.2011.08.033; PMID: 21982369
- Luk KC, Kehm VM, Zhang B, O’Brien P, Trojanowski JQ, Lee VM. Intracerebral inoculation of pathological α-synuclein initiates a rapidly progressive neurodegenerative α-synucleinopathy in mice. J Exp Med 2012; 209:975 - 86; http://dx.doi.org/10.1084/jem.20112457; PMID: 22508839
- Masuda-Suzukake M, Nonaka T, Hosokawa M, Oikawa T, Arai T, Akiyama H, Mann DM, Hasegawa M. Prion-like spreading of pathological α-synuclein in brain. Brain 2013; 136:1128 - 38; http://dx.doi.org/10.1093/brain/awt037; PMID: 23466394
- Sacino AN, Brooks M, McGarvey NH, McKinney AB, Thomas MA, Levites Y, Ran Y, Golde TE, Giasson BI. Induction of CNS α-synuclein pathology by fibrillar and non-amyloidogenic recombinant α-synuclein. Acta Neuropathol Commun 2013; 1:38; http://dx.doi.org/10.1186/2051-5960-1-38; PMID: 24252149
- Aguzzi A, Heikenwalder M, Polymenidou M. Insights into prion strains and neurotoxicity. Nat Rev Mol Cell Biol 2007; 8:552 - 61; http://dx.doi.org/10.1038/nrm2204; PMID: 17585315
- Ren PH, Lauckner JE, Kachirskaia I, Heuser JE, Melki R, Kopito RR. Cytoplasmic penetration and persistent infection of mammalian cells by polyglutamine aggregates. Nat Cell Biol 2009; 11:219 - 25; http://dx.doi.org/10.1038/ncb1830; PMID: 19151706
- Frost B, Ollesch J, Wille H, Diamond MI. Conformational diversity of wild-type Tau fibrils specified by templated conformation change. J Biol Chem 2009; 284:3546 - 51; http://dx.doi.org/10.1074/jbc.M805627200; PMID: 19010781
- Lee HJ, Suk JE, Bae EJ, Lee JH, Paik SR, Lee SJ. Assembly-dependent endocytosis and clearance of extracellular alpha-synuclein. Int J Biochem Cell Biol 2008; 40:1835 - 49; http://dx.doi.org/10.1016/j.biocel.2008.01.017; PMID: 18291704
- Luk KC, Song C, O’Brien P, Stieber A, Branch JR, Brunden KR, Trojanowski JQ, Lee VM. Exogenous alpha-synuclein fibrils seed the formation of Lewy body-like intracellular inclusions in cultured cells. Proc Natl Acad Sci U S A 2009; 106:20051 - 6; http://dx.doi.org/10.1073/pnas.0908005106; PMID: 19892735
- Danzer KM, Krebs SK, Wolff M, Birk G, Hengerer B. Seeding induced by alpha-synuclein oligomers provides evidence for spreading of alpha-synuclein pathology. J Neurochem 2009; 111:192 - 203; http://dx.doi.org/10.1111/j.1471-4159.2009.06324.x; PMID: 19686384
- Steiner JA, Angot E, Brundin P. A deadly spread: cellular mechanisms of α-synuclein transfer. Cell Death Differ 2011; 18:1425 - 33; http://dx.doi.org/10.1038/cdd.2011.53; PMID: 21566660
- Lee HJ, Patel S, Lee SJ. Intravesicular localization and exocytosis of alpha-synuclein and its aggregates. J Neurosci 2005; 25:6016 - 24; http://dx.doi.org/10.1523/JNEUROSCI.0692-05.2005; PMID: 15976091
- Freundt EC, Maynard N, Clancy EK, Roy S, Bousset L, Sourigues Y, Covert M, Melki R, Kirkegaard K, Brahic M. Neuron-to-neuron transmission of α-synuclein fibrils through axonal transport. Ann Neurol 2012; 72:517 - 24; http://dx.doi.org/10.1002/ana.23747; PMID: 23109146
- Sherer NM, Mothes W. Cytonemes and tunneling nanotubules in cell-cell communication and viral pathogenesis. Trends Cell Biol 2008; 18:414 - 20; http://dx.doi.org/10.1016/j.tcb.2008.07.003; PMID: 18703335
- Gousset K, Schiff E, Langevin C, Marijanovic Z, Caputo A, Browman DT, Chenouard N, de Chaumont F, Martino A, Enninga J, et al. Prions hijack tunnelling nanotubes for intercellular spread. Nat Cell Biol 2009; 11:328 - 36; http://dx.doi.org/10.1038/ncb1841; PMID: 19198598
- Kayed R, Head E, Thompson JL, McIntire TM, Milton SC, Cotman CW, Glabe CG. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science 2003; 300:486 - 9; http://dx.doi.org/10.1126/science.1079469; PMID: 12702875
- Lashuel HA, Overk CR, Oueslati A, Masliah E. The many faces of α-synuclein: from structure and toxicity to therapeutic target. Nat Rev Neurosci 2013; 14:38 - 48; http://dx.doi.org/10.1038/nrn3406; PMID: 23254192
- Winner B, Jappelli R, Maji SK, Desplats PA, Boyer L, Aigner S, Hetzer C, Loher T, Vilar M, Campioni S, et al. In vivo demonstration that alpha-synuclein oligomers are toxic. Proc Natl Acad Sci U S A 2011; 108:4194 - 9; http://dx.doi.org/10.1073/pnas.1100976108; PMID: 21325059
- Martin ZS, Neugebauer V, Dineley KT, Kayed R, Zhang W, Reese LC, Taglialatela G. α-Synuclein oligomers oppose long-term potentiation and impair memory through a calcineurin-dependent mechanism: relevance to human synucleopathic diseases. J Neurochem 2012; 120:440 - 52; http://dx.doi.org/10.1111/j.1471-4159.2011.07576.x; PMID: 22060133
- Volles MJ, Lee SJ, Rochet JC, Shtilerman MD, Ding TT, Kessler JC, Lansbury PT Jr.. Vesicle permeabilization by protofibrillar alpha-synuclein: implications for the pathogenesis and treatment of Parkinson’s disease. Biochemistry 2001; 40:7812 - 9; http://dx.doi.org/10.1021/bi0102398; PMID: 11425308
- Perrin RJ, Woods WS, Clayton DF, George JM. Interaction of human alpha-Synuclein and Parkinson’s disease variants with phospholipids. Structural analysis using site-directed mutagenesis. J Biol Chem 2000; 275:34393 - 8; http://dx.doi.org/10.1074/jbc.M004851200; PMID: 10952980
- Kayed R, Sokolov Y, Edmonds B, McIntire TM, Milton SC, Hall JE, Glabe CG. Permeabilization of lipid bilayers is a common conformation-dependent activity of soluble amyloid oligomers in protein misfolding diseases. J Biol Chem 2004; 279:46363 - 6; http://dx.doi.org/10.1074/jbc.C400260200; PMID: 15385542
- Glabe CG. Common mechanisms of amyloid oligomer pathogenesis in degenerative disease. Neurobiol Aging 2006; 27:570 - 5; http://dx.doi.org/10.1016/j.neurobiolaging.2005.04.017; PMID: 16481071
- Cremades N, Cohen SIA, Deas E, Abramov AY, Chen AY, Orte A, Sandal M, Clarke RW, Dunne P, Aprile FA, et al. Direct observation of the interconversion of normal and toxic forms of α-synuclein. Cell 2012; 149:1048 - 59; http://dx.doi.org/10.1016/j.cell.2012.03.037; PMID: 22632969
- Pieri L, Madiona K, Bousset L, Melki R. Fibrillar α-synuclein and huntingtin exon 1 assemblies are toxic to the cells. Biophys J 2012; 102:2894 - 905; http://dx.doi.org/10.1016/j.bpj.2012.04.050; PMID: 22735540
- Watts JC, Giles K, Oehler A, Middleton L, Dexter DT, Gentleman SM, DeArmond SJ, Prusiner SB. Transmission of multiple system atrophy prions to transgenic mice. Proc Natl Acad Sci U S A 2013; 110:19555 - 60; http://dx.doi.org/10.1073/pnas.1318268110; PMID: 24218576
- Liu IH, Uversky VN, Munishkina LA, Fink AL, Halfter W, Cole GJ. Agrin binds alpha-synuclein and modulates alpha-synuclein fibrillation. Glycobiology 2005; 15:1320 - 31; http://dx.doi.org/10.1093/glycob/cwj014; PMID: 16037493
- Giehm L, Oliveira CLP, Christiansen G, Pedersen JS, Otzen DE. SDS-induced fibrillation of alpha-synuclein: an alternative fibrillation pathway. J Mol Biol 2010; 401:115 - 33; http://dx.doi.org/10.1016/j.jmb.2010.05.060; PMID: 20540950
- Uversky VN, Li J, Souillac P, Millett IS, Doniach S, Jakes R, Goedert M, Fink AL. Biophysical properties of the synucleins and their propensities to fibrillate: inhibition of alpha-synuclein assembly by beta- and gamma-synucleins. J Biol Chem 2002; 277:11970 - 8; http://dx.doi.org/10.1074/jbc.M109541200; PMID: 11812782
- Li J, Zhu M, Manning-Bog AB, Di Monte DA, Fink AL. Dopamine and L-dopa disaggregate amyloid fibrils: implications for Parkinson’s and Alzheimer’s disease. FASEB J 2004; 18:962 - 4; PMID: 15059976
- Zhu M, Fink AL. Lipid binding inhibits alpha-synuclein fibril formation. J Biol Chem 2003; 278:16873 - 7; http://dx.doi.org/10.1074/jbc.M210136200; PMID: 12621030
- Li J, Zhu M, Rajamani S, Uversky VN, Fink AL. Rifampicin inhibits alpha-synuclein fibrillation and disaggregates fibrils. Chem Biol 2004; 11:1513 - 21; http://dx.doi.org/10.1016/j.chembiol.2004.08.025; PMID: 15556002
- Jiang T, Yu WB, Yao T, Zhi XL, Pan LF, Wang J, et al. Trehalose inhibits wild-type alpha-synuclein fibrillation and overexpression and protects against the protein neurotoxicity in transduced PC12 cells. Rsc Advances 2013; 3:9500 - 8; http://dx.doi.org/10.1039/c3ra40600h
- Uversky V. α-Synuclein Aggregation and Parkinson’s Disease. In: Uversky V, Fink A, eds. Protein Misfolding, Aggregation, and Conformational Diseases: Springer US, 2007:61-110.
- Chavarría C, Souza JM. Oxidation and nitration of α-synuclein and their implications in neurodegenerative diseases. Arch Biochem Biophys 2013; 533:25 - 32; http://dx.doi.org/10.1016/j.abb.2013.02.009; PMID: 23454347
- Moriarty GM, Janowska MK, Kang L, Baum J. Exploring the accessible conformations of N-terminal acetylated α-synuclein. FEBS Lett 2013; 587:1128 - 38; http://dx.doi.org/10.1016/j.febslet.2013.02.049; PMID: 23499431