
Abstract
Beta amyloid (Aβ) accumulation is recognized as a hallmark of Alzheimer disease (AD) pathology and the aggregation of Aβ peptide is hypothesized to drive pathogenesis. As such, Aβ is a logical target for therapeutic intervention and there have been many studies looking at diverse classes of drugs that target Aβ. Of concern is the recent failure of several clinical trials, highlighting the need for earlier, possibly preventative intervention, and raising the question of what form of Aβ is the best target. The Aβ oligomers are considered to be the toxic species, but many therapies, such as antibody therapies, target monomers, removing them as substrates for aggregation. Peptide inhibitors, in contrast, are able to interfere with the aggregation process itself. Designing peptide inhibitors requires some knowledge of Aβ structure; while there is structural information about the amyloid core of Aβ fibrils, the transient nature of oligomers makes them difficult to characterize. Fortunately, some interaction sites have been identified between monomers and oligomers of Aβ and these, plus known aggregation-prone sequences in Aβ, can serve as a basis for inhibitor design. In this mini-review we focus on D-amino acid based peptide inhibitors and discuss how their non-toxic and stable nature can be beneficial, while they specifically target aggregation-prone sequences within the Aβ peptide. Many peptide inhibitors have been designed using the LVFFA domain within Aβ to disrupt the self-assembly of Aβ peptide. While this may be sufficient to stop aggregation in vitro, other aggregation sites at the C-terminus may promote aggregation independently and the flexible N terminus may be a good target to induce clearance of aggregates. Ultimately, it may be a combination of targets that provides the best therapeutic strategy.
Introduction
Alzheimer disease (AD) is the sixth major cause of death in the US and the only one among the top ten that lacks a means of prevention, cure or disease-modifying treatment.Citation1,Citation2 AD is the most common form of dementia and it causes a gradually progressive decline in cognitive abilities. Unfortunately, clinical trials have not been successful in reducing or preventing AD. In order to understand the failure of these trials, one has to understand how the chosen therapeutic targets relate to the pathogenesis of disease and how timing of intervention may be critical.
Most therapies have targeted the β-amyloid (Aβ) cascade; according to the Aβ cascade hypothesis,Citation3 it is the conformational transformation of Aβ peptide which triggers AD pathology. This is not to suggest that Aβ is the only misfolded protein in AD pathogenesis; neurofibrillary tangles (NFTs) of hyperphoshorylated tau also occur and contribute to synaptic degeneration and neuronal cell loss, but NFTs are thought to arise because of Aβ oligomerization. The Aβ peptide forms as a result of proteolysis of amyloid precursor protein (APP). It can be 39–43 residues long but Aβ1-42 is the most abundant form present in the brains of AD patients. Aβ monomers undergo a conformational change and become aggregation-prone. They quickly arrange themselves into transient oligomeric aggregates of variable sizes and ultimately produce fibrils and amyloid plaques in the brain. Both the Aβ oligomers and fibrils are known to be neurotoxic.
In addition to the appropriate therapeutic target, timing of treatment is important. By the time a diagnosis of AD is made and treatment can begin, there is already significant plaque burden; preventing further plaque accumulation at this stage may be too late, as seen in a study of solanezumab, an antibody against Aβ which prevents plaque formation. In patients with moderate disease, it did not slow progression, but in patients with mild deficits, cognitive decline was slowed.Citation4 This emphasizes the need to intervene at an earlier stage of the cascade, when Aβ is starting to aggregate into oligomers. Ideally, treatment would be preventative, starting long before patients develop signs or symptoms, and would reduce pathological forms of Aβ, particularly oligomers. For most patients, we cannot yet predict who will develop AD, but prevention studies are underway in the subset of patients with genetic AD, using approaches that target monomeric Aβ with antibodies (crenezumab and solanezumab) and that reduce Aβ production by inhibiting the β-secretase (BACE) that produces Aβ from APP (such as minocycline). Toxicity can be an issues in such trials; a study of BACE inhibitor LY2886721 was halted due to liver abnormalities.Citation5 Nevertheless, these early intervention studies will help confirm whether early targeting of the Aβ cascade is a viable therapeutic approach.
Another class of AD therapeutic that targets the Aβ cascade is the peptide inhibitor. Whereas antibodies target Aβ monomers, thus removing the substrate for aggregation, peptide inhibitors specifically interfere with Aβ assembly. Some of these inhibitors have also entered into clinical trials. In this mini-review we discuss the rationale for targeting the aggregation process itself and the evidence for a specific subset of peptide therapy that uses D-amino acids, as these amino acids are non-toxic and improve the stability of peptides. Ultimately, it may be that a combination of therapeutic targets within the Aβ cascade will yield the best clinical outcomes.
Targeting Aβ Structural Signatures: Monomers vs. Oligomers vs. Fibrils
Because of the heterogeneity of aggregating Aβ peptides, it is difficult to generate antibodies against a toxic species directly. Instead, most antibodies target and bind the monomeric form of the peptide in its helical form, not its β-sheet rich disease-associated form.Citation6,Citation7 This serves to remove the substrate from the aggregation process. In contrast, an effective peptide inhibitor should be designed based on structural signatures of the parent peptide in its disease-associated form(s). In AD, this raises the question of which disease-associated aggregate structure to choose, as many aggregates are already formed by the time clinical symptoms present ().
Figure 1. Schematic diagram of Aβ aggregation in the context of Alzheimer disease progression. The diagram is based on in vitro aggregation data and in vivo findings of Aβ plaque formation. The disease has a prolonged preclinical state, in which Aβ peptides undergo major structural transitions to form oligomers then fibrils. These fibrils undergo continuous fragmentation and reassembly during the course of the disease.Citation46
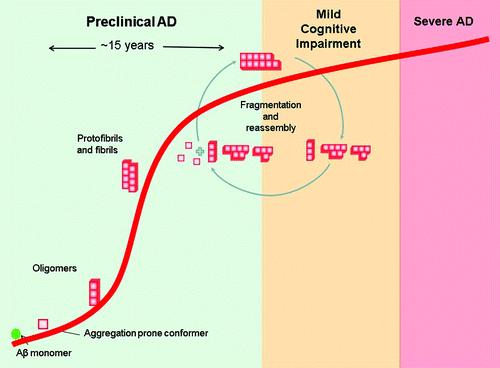
Studies of the aggregation process indicate that native Aβ peptides undergo a structural transition and form transient monomers/oligomers. Some of these transient molecules can lead to the formation of low molecular weight (LMW) oligomers, which vary in size and shape. These LMW oligomers in turn tend to form large aggregate assemblies known as protofibrils or high molecular weight (HMW) oligomers. Ultimately, these protofibrils form amyloid plaques. The steps involved in the formation of amyloid plaques are reversible in nature but favor plaque formation once LMW oligomers are formed.
The exact pathways of aggregation and corresponding structures have been studied by Eisenberg and his colleagues, who have suggested the existence of two distinct aggregation pathways for amyloid forming proteins like Aβ.Citation8 These pathways have different energy barriers and depend on the length of the starting peptide. For Aβ1–40 and 1–42, a fibril pathway led to the formation of in-register β-sheet fibers as observed by solid state nuclear magnetic resonance (NMR) spectroscopy. In these structures, β-strands were on top of or underneath a similar β-strand.
Using shorter Aβ peptides (Aβ16–21, 25–35, 30–36), a second pathway was observed, resulting in out-of-register β-sheet oligomers in which antiparallel β-strands were observed. Interestingly, these out-of-register oligomers were cytotoxic.Citation8,Citation9 Whether these cytotoxic oligomers reflect what occurs in vivo is of particular interest; over the last decade, Aβ oligomers that have been recognized as the most toxic species, but it is not clear whether they are on or off-pathway to plaque formation or are oligomers shed from mature fibrils.Citation10-Citation16
Aggregation-Prone Sequences and Interaction Sites In Aβ
The Aβ peptide is an aggregation prone peptide, with its LVFFACitation17 and GxxxG motifs being identified as particularly aggregation-prone based on a number of studies. Within the Aβ sequence, GxxxG and GxxxA are reported to be transmembrane dimerization motifsCitation18 and a number of familial mutations lie within the GxxxG motifs. The Glycine at residue 33 plays a critical role in early aggregation of Aβ42 into oligomersCitation19 and a peptide inhibitor (RGTFEGKF) that targets the GxxxG motif has been shown to disrupt the formation of mature Aβ fibrils.Citation20
Our understanding of aggregation is largely based on the amyloid structures solved using solid state NMR, as described above. These studies have revealed that Aβ1–42 fibrils are assembled as in-register parallel β-sheets with hydrophobic interactions occurring among L17, F19, A21, A30, I32, L34, and V36 residues.Citation21,Citation22 However, the study of fibrillar amyloid does not necessarily reveal how the initial aggregation steps occur between monomer-monomer or monomer-oligomer, and solid state NMR methods are unable to characterize transient oligomer species. The best that can be proposed is that an Aβ monomer may bind the hydrophobic residues 17–21, be partially stabilized by intermolecular salt-bridge and backbone H bonds, and then become fully stabilized with the addition of the next monomer.Citation22 This sequence-selective, cooperative mechanism of fibril growth follows first-order kinetics but does not take into account any oligomeric intermediates.
Fortunately, liquid NMR can allow an indirect study of the structural transition that occurs when Aβ peptide forms oligomers. Fawzi et al.Citation23 used exchange NMR to study Aβ monomer-oligomer interactions. They observed labeled monomers interacting with unlabelled oligomers and protofibrils. They determined that Aβ sequences LVFFA (17–21), AII (30–32), LMV (34–36) and VVIA (39–42) were the initial contact points between Aβ monomers and oligomers (). Notably, apart from residues 39–42, all these regions have hydrophobic interactions within the amyloid form, suggesting that these initial interactions may indeed remain throughout the fibrillization process. In addition, both the AII (30–32) and LMV (34–36) interaction sites lie within a GxxxG motif.
Figure 2. Aβ1–42 peptide showing monomer-oligomer interaction sites. (A) Aβ1–42 sequence. Monomeric Aβ has been shown to interact with oligomeric forms of Aβ at the residues indicated in red.Citation23 (B) Three dimensional structure of Aβ1–42 based on liquid NMR. Interaction sites between monomer and oligomer are again displayed in red within the three dimensional monomer.Citation47 (C) Three dimensional structure of amyloid Aβ1–42 based on solid state NMR, showing core region β sheet residues 17–42.Citation22 The monomer-oligomer interaction sites are again shown in red with known hydrophobic interactions demonstrated as side chains. Residues 1–16 remain as a flexible N-terminus in this amyloid model and so the three N-terminal sites of oligomer interaction cannot be mapped onto this model. The remaining oligomer interaction sites are all within the amyloid core region, wih the highest correlation being between the two main aggregation motifs LVFFA and GxxxG.
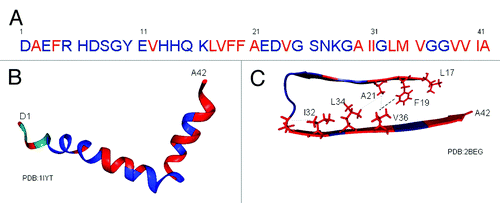
Fawzi et al.Citation23 also observed oligomer associations at several single amino acid residues (A2, F4, V12, V24), although their proximity interaction was less significant. This was interesting in that N-terminal interactions had not been observed by solid-state NMR spectroscopy of fibrils because this region is mobile and flexible, remaining outside the amyloid core. A recent study of Aβ1–40 has also found interactions between the monomer and Aβ1–40 aggregates occurring at N-terminal residues.Citation24
Taken together, the solid state and liquid NMR studies indicate that there is more than one aggregation-prone region within the Aβ sequence, and that N-terminal residues may play a role in initial interactions between monomers and oligomers. As such, all sites are potential targets for peptide inhibitor design.
D-Amino Acid Peptides as Drug Therapy
As early as in 1992, Beyreuther and colleaguesCitation17 demonstrated that replacing hydrophobic residues within the Aβ peptide reduced its amyloidogenicity, which is in keeping with current knowledge regarding potential initiation sites or self recognition elements (SREs) within the Aβ sequence. They also showed that in vitro fibril properties were the same for full-length, 10–43 or 10–23 and that substitutions at positions 17–20 were sufficient to reduce amyloidogenicity. Since then, a number of studies have been performed which target part or all of the amyloidogenic sequence HQKLVFFAE (15–22) using D-amino acid substitutions.
D-Amino acids are the stereoisomers of naturally occurring L-amino acids and are considered non-natural amino acids. Some prokaryotes produce and use D-amino acids and they can be found in the cell walls of bacteria, but generally D-amino acids are only produced during chemical synthesis. Peptides containing D-amino acids can mimic naturally occurring peptides, but they are more protease resistant, which makes them more suitable for therapeutics.Citation25 For example, Icatibant, a bradykinin B2 receptor antagonist decapeptide, is an FDA-approved treatment for hereditary angioedema that contains five non-natural amino acids. The D-amino acids prevent Icatibant from being metabolized by major bradykinin-metabolizing enzymes.Citation26 Another example is the retro-inverso peptide RI-OR2, which targets the Aβ peptide (discussed later), and has a prolonged half-life when compared with its L-amino acid counterpart.Citation27
In the following discussion, amino acids which are in the D isomer configuration are written in lower case.
Preventing aggregation
Logical outcomes for screening peptide inhibitors are the abilities to prevent aggregation and prevent toxicity. In a study by Soto et al.,Citation28 it was shown that an 11 residue long D-peptide rdlpffpvpid (D-iAβ1) had a similar inhibitory effect as the L-peptide version (L-iAβ1) on fibril formation of full-length Aβ peptide, but D-iAβ1 is reported to be more stable against proteolysis, although it has not been tested in a cellular model.
Shorter peptides from within this sequence can also have inhibitory effects. The L-peptide KLVFF (16–20) can prevent the formation of Aβ1–40 fibersCitation29 and, according to in-silico modeling, it binds to the homologous sequence on Aβ peptide. This peptide also assembles itself into Thioflavin T (ThT) negative fibrils. While the D-peptide version (klvff) was unable to bind Aβ13–22,Citation30 a similar D-pentapeptide (cholyl-lvffa) inhibited fibril formation and cytotoxicity.Citation31
A screen of combinatorial pentapeptide libraries composed of D-amino acids identified a number of peptides containing phenylalanine in the second position followed by leucine at the third. Those D-pentapeptide ligands were capable of binding to Aβ and also prevented formation of fibrils.Citation29
In 2003, Chalifour et al. systematically analyzed the inhibitory effects of hexa and heptapeptides from the Aβ sequence and found that the peptides containing D-enantiomers for residues 15–20 were most effective, followed by 16–22, which clearly identified residues 16–20 KLVFF as a target motif for D-peptides. The D-peptides klvffa, kklvffa, kfvffa, kivffa and kvvffa were studied in detail; klvffa was the most effective at inhibiting fibril formation and kklvffa was more effective in rescuing cellular toxicity in SH-SY5Y cells. In another experiment, a single residue D- to L-amino acid substitution within klvffa reduced inhibitory effects most significantly when V18 and F19 were switched to their L-enantiomer.Citation32
Promoting the formation of stable, non-toxic, off-pathway aggregates
Another potential mechanism of action for a peptide inhibitor is to promote the aggregation of stable non-toxic aggregates. Recent studiesCitation33,Citation34 have shown that formation of stable Aβ aggregates reduces cytotoxicity. There are also several studies in which cytoprotection has been observed in association with the formation of large off-pathway aggregates.
The group of Willbold in Germany, used a mirror image page display method to identify the 12 residue D-amino acid peptide D-pep (qshyrhispaqv) as a potential inhibitor. D-pep was shown to specifically bind Aβ fibrils contained in plaques and leptomeningeal vessels.Citation35 D-pep reduced the Aβ particle size but actually increased the overall number of aggregates; despite this, it also increased cell viability in PC12 cells as measured by MTT assay.Citation36
The same group then identified another D-Peptide D3 (rprtrlhthrnr), which had the ability to promote disassembly of preformed Aβ aggregates, to increase cell viability in PC12 cells and to reduce the amyloid plaque burden in APP/PS1 double transgenic mice.Citation37 They demonstrated that these D-peptides, when injected or infused into animal, did not cause an inflammatory response and were able to bind to Aβ1–42 in the brains of transgenic mice. They also found that D3 was taken up by neurons.Citation38 Oral treatment with D3 led to an improvement in pathology and behavior in this mouse model and indicated that the peptide was able to cross blood brain barrier using transcytosis.Citation39 Dynamic light scattering data indicated that the D3 peptide caused the Aβ peptide to form a distinct population of aggregates with hydrodynamic radii of 800 nm, suggesting that D3 peptide does not prevent aggregation, but drives Aβ to form amorphous aggregates which are less toxic.
Doig et al.Citation40 also explored the structure-activity relationship of a series of peptides targeting KLVFFA in the Aβ sequence. In addition to using D-amino acids, they introduced N-methylation. They identified SEN304 (cg-y-cg-cg-ml), where all the glycine residues had a cyclohexyl group added and the leucine was methylated, as the most effective peptide against aggregation. They characterized SEN304 further using various biophysical assays along with toxicity assays in SH-SHY5Y cells, neuronal viability and synaptophysin levels in mouse neuronal cultures, and long-term potentiation in rat hippocampal brain slices.Citation41 The inhibition of long-term potentiation by 1 μM Aβ1–42 was reversed by adding 100 nM SEN304 to the rat hippocampal brain slices. Electron microscopic analysis supported their conclusion, as seen with the other D-peptides, that SEN304 drives Aβ monomer and oligomers to alternative large ThT-negative aggregates that are not cytotoxic.
In vivo studies
A true inhibitor needs to be effective in animal models, not just cell culture. Most of the studied D amino acid peptides are too large to cross the blood brain barrier (BBB) and we must remember that AD related structural transition and aggregation happens in the brain. Therefore, the BBB is the biggest hindrance to the delivery of peptide-based therapeutics in AD. The BBB limits drug size to approximately 1 nm, unless modified by using receptors involved in the transcytosis of ligands from the blood to the brain. These modifications can be achieved by protein engineering using HIV1-TAT or by addition of PEG for increased solubility.
In addition to the Willbold studies of D-3 mentioned above, several other groups have tested inhibitory D-peptides in animal models. The retro-inverso peptide RI-OR2 (rGklvffGr) has been shown to be a more effective anti-aggregation peptide than OR2 which consists of same amino acids in L configuration.Citation27,Citation42 This peptide acts on Aβ oligomers as well as on fibril formation and has protective effects on SH-SY5Y cells. The HIV-1 TAT sequence was introduced to the peptide to allow transport through the BBB; this did not change the properties RI-OR2 in the context of Aβ aggregation. The RI-OR2-TAT peptides were able to penetrate the BBB and reduce the number of activated microglia in transgenic APP/PS1 mouse brains.Citation43
Gazit et al. studied a small non-coded amino acid α-aminoisobutyric acid (Aib) as a small dipeptide with D-Trp. The D-Trp-Aib targets aromatic interfaces in Aβ peptide and can reduce the amyloid deposit in AD transgenic mice.Citation44
Expanding the Target Region
While D-amino acid peptides have advantages of protease resistance and have been proven effective against the central LVFFA region of Aβ, there are other regions that may also be good targets. The Aβ peptide can be considered as having three domains: a flexible N-terminus, which remains outside the amyloid core, and two arms of the core. The N-terminus, which is flexible in all structures studied, may be involved in early interactions with oligomersCitation23 and may also be used to enhance clearance.Citation45 Of the arms of the core, one contains the aggregation-prone LVFFA sequence and the other has GxxxG motifs, where the middle residues are AII (30–32) and LMV (34–36) (). The aggregation-prone Aβ monomers / oligomers likely undergo self adhesion through different SREs in vivo; at any given time, the peptide may be present in a number of different conformational states. Fortunately, the central LVFFA, GxxxG motifs and N-terminus are accessible in all the known amyloid structures, so these targets are likely available to drugs at many if not all stages of aggregation. Targeting both the LVFFA and GxxxG regions simultaneously could provide synergistic effects on aggregation, either leading to the complete inhibition of aggregation or the promotion of stable off-pathway oligomers which are non-toxic. The role of the flexible N-terminus within Aβ amyloid plaques is not well understood, but its solvent accessibility may be used to facilitate the clearance of any aggregates which do form.
Conclusion
Alzheimer disease is rapidly becoming a major health and socioeconomic concern for our society. The protein misfolding which underlies this disease provides several targets for possible early therapeutic intervention. D-amino acid peptides are one such therapeutic option. They can have cytoprotective effects with short sequence lengths that can cross the blood brain barrier, they have selectivity for Aβ, and they have a greater protease resistance than their L-amino acid counterparts. By using combinations of sequence targets, both aggregation and clearance may be influenced. They are one of most suitable candidates for preventive therapy. As such, there is a role for further study of these peptides.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Related Research Data
References
- http://www.alz.org/documents_custom/2013_facts_figures_fact_sheet.pdf.
- http://www.cdc.gov/nchs/data/databriefs/db116.htm.
- Hardy JA, Higgins GA. Alzheimer’s disease: the amyloid cascade hypothesis. Science 1992; 256:184 - 5; http://dx.doi.org/10.1126/science.1566067; PMID: 1566067
- Callaway E. Alzheimer’s drugs take a new tack. Nature 2012; 489:13 - 4; http://dx.doi.org/10.1038/489013a; PMID: 22962697
- https://investor.lilly.com/releasedetail.cfm?ReleaseID=771353.
- Miles LA, Crespi GA, Doughty L, Parker MW. Bapineuzumab captures the N-terminus of the Alzheimer’s disease amyloid-beta peptide in a helical conformation. Sci Rep 2013; 3:1302; PMID: 23416764
- Racke MM, Boone LI, Hepburn DL, Parsadainian M, Bryan MT, Ness DK, Piroozi KS, Jordan WH, Brown DD, Hoffman WP, et al. Exacerbation of cerebral amyloid angiopathy-associated microhemorrhage in amyloid precursor protein transgenic mice by immunotherapy is dependent on antibody recognition of deposited forms of amyloid beta. J Neurosci 2005; 25:629 - 36; http://dx.doi.org/10.1523/JNEUROSCI.4337-04.2005; PMID: 15659599
- Liu C, Zhao M, Jiang L, Cheng PN, Park J, Sawaya MR, Pensalfini A, Gou D, Berk AJ, Glabe CG, et al. Out-of-register β-sheets suggest a pathway to toxic amyloid aggregates. Proc Natl Acad Sci U S A 2012; 109:20913 - 8; http://dx.doi.org/10.1073/pnas.1218792109; PMID: 23213214
- Laganowsky A, Liu C, Sawaya MR, Whitelegge JP, Park J, Zhao M, Pensalfini A, Soriaga AB, Landau M, Teng PK, et al. Atomic view of a toxic amyloid small oligomer. Science 2012; 335:1228 - 31; http://dx.doi.org/10.1126/science.1213151; PMID: 22403391
- Hartley DM, Walsh DM, Ye CP, Diehl T, Vasquez S, Vassilev PM, Teplow DB, Selkoe DJ. Protofibrillar intermediates of amyloid beta-protein induce acute electrophysiological changes and progressive neurotoxicity in cortical neurons. J Neurosci 1999; 19:8876 - 84; PMID: 10516307
- Cleary JP, Walsh DM, Hofmeister JJ, Shankar GM, Kuskowski MA, Selkoe DJ, Ashe KH. Natural oligomers of the amyloid-beta protein specifically disrupt cognitive function. Nat Neurosci 2005; 8:79 - 84; http://dx.doi.org/10.1038/nn1372; PMID: 15608634
- Silveira JR, Raymond GJ, Hughson AG, Race RE, Sim VL, Hayes SF, Caughey B. The most infectious prion protein particles. Nature 2005; 437:257 - 61; http://dx.doi.org/10.1038/nature03989; PMID: 16148934
- Xue WF, Hellewell AL, Gosal WS, Homans SW, Hewitt EW, Radford SE. Fibril fragmentation enhances amyloid cytotoxicity. J Biol Chem 2009; 284:34272 - 82; http://dx.doi.org/10.1074/jbc.M109.049809; PMID: 19808677
- Cremades N, Cohen SI, Deas E, Abramov AY, Chen AY, Orte A, Sandal M, Clarke RW, Dunne P, Aprile FA, et al. Direct observation of the interconversion of normal and toxic forms of α-synuclein. Cell 2012; 149:1048 - 59; http://dx.doi.org/10.1016/j.cell.2012.03.037; PMID: 22632969
- Krishnan R, Goodman JL, Mukhopadhyay S, Pacheco CD, Lemke EA, Deniz AA, Lindquist S. Conserved features of intermediates in amyloid assembly determine their benign or toxic states. Proc Natl Acad Sci U S A 2012; 109:11172 - 7; http://dx.doi.org/10.1073/pnas.1209527109; PMID: 22745165
- Shahnawaz M, Soto C. Microcin amyloid fibrils A are reservoir of toxic oligomeric species. J Biol Chem 2012; 287:11665 - 76; http://dx.doi.org/10.1074/jbc.M111.282533; PMID: 22337880
- Hilbich C, Kisters-Woike B, Reed J, Masters CL, Beyreuther K. Substitutions of hydrophobic amino acids reduce the amyloidogenicity of Alzheimer’s disease beta A4 peptides. J Mol Biol 1992; 228:460 - 73; http://dx.doi.org/10.1016/0022-2836(92)90835-8; PMID: 1453457
- Brosig B, Langosch D. The dimerization motif of the glycophorin A transmembrane segment in membranes: importance of glycine residues. Protein Sci 1998; 7:1052 - 6; http://dx.doi.org/10.1002/pro.5560070423; PMID: 9568912
- Harmeier A, Wozny C, Rost BR, Munter LM, Hua H, Georgiev O, Beyermann M, Hildebrand PW, Weise C, Schaffner W, et al. Role of amyloid-beta glycine 33 in oligomerization, toxicity, and neuronal plasticity. J Neurosci 2009; 29:7582 - 90; http://dx.doi.org/10.1523/JNEUROSCI.1336-09.2009; PMID: 19515926
- Sato T, Kienlen-Campard P, Ahmed M, Liu W, Li H, Elliott JI, Aimoto S, Constantinescu SN, Octave JN, Smith SO. Inhibitors of amyloid toxicity based on beta-sheet packing of Abeta40 and Abeta42. Biochemistry 2006; 45:5503 - 16; http://dx.doi.org/10.1021/bi052485f; PMID: 16634632
- Balbach JJ, Petkova AT, Oyler NA, Antzutkin ON, Gordon DJ, Meredith SC, Tycko R. Supramolecular structure in full-length Alzheimer’s beta-amyloid fibrils: evidence for a parallel beta-sheet organization from solid-state nuclear magnetic resonance. Biophys J 2002; 83:1205 - 16; http://dx.doi.org/10.1016/S0006-3495(02)75244-2; PMID: 12124300
- Lührs T, Ritter C, Adrian M, Riek-Loher D, Bohrmann B, Döbeli H, Schubert D, Riek R. 3D structure of Alzheimer’s amyloid-beta(1-42) fibrils. Proc Natl Acad Sci U S A 2005; 102:17342 - 7; http://dx.doi.org/10.1073/pnas.0506723102; PMID: 16293696
- Fawzi NL, Ying J, Ghirlando R, Torchia DA, Clore GM. Atomic-resolution dynamics on the surface of amyloid-β protofibrils probed by solution NMR. Nature 2011; 480:268 - 72; http://dx.doi.org/10.1038/nature10577; PMID: 22037310
- Ono K, et al. Exogenous amyloidogenic proteins function as seeds in amyloid β-protein aggregation. Biochimica et Biophysica Acta (BBA) -. Molecular Basis of Disease 2014; 1842:646 - 53; http://dx.doi.org/10.1016/j.bbadis.2014.01.002
- Zhou N, Luo Z, Luo J, Fan X, Cayabyab M, Hiraoka M, Liu D, Han X, Pesavento J, Dong CZ, et al. Exploring the stereochemistry of CXCR4-peptide recognition and inhibiting HIV-1 entry with D-peptides derived from chemokines. J Biol Chem 2002; 277:17476 - 85; http://dx.doi.org/10.1074/jbc.M202063200; PMID: 11880384
- Bork K, Yasothan U, Kirkpatrick P. Icatibant. Nat Rev Drug Discov 2008; 7:801 - 2; http://dx.doi.org/10.1038/nrd2694
- Taylor M, Moore S, Mayes J, Parkin E, Beeg M, Canovi M, Gobbi M, Mann DM, Allsop D. Development of a proteolytically stable retro-inverso peptide inhibitor of beta-amyloid oligomerization as a potential novel treatment for Alzheimer’s disease. Biochemistry 2010; 49:3261 - 72; http://dx.doi.org/10.1021/bi100144m; PMID: 20230062
- Soto C, Kindy MS, Baumann M, Frangione B. Inhibition of Alzheimer’s amyloidosis by peptides that prevent beta-sheet conformation. Biochem Biophys Res Commun 1996; 226:672 - 80; http://dx.doi.org/10.1006/bbrc.1996.1413; PMID: 8831674
- Tjernberg LO, Näslund J, Lindqvist F, Johansson J, Karlström AR, Thyberg J, Terenius L, Nordstedt C. Arrest of beta-amyloid fibril formation by a pentapeptide ligand. J Biol Chem 1996; 271:8545 - 8; http://dx.doi.org/10.1074/jbc.271.15.8545; PMID: 8621479
- Tjernberg LO, Lilliehöök C, Callaway DJ, Näslund J, Hahne S, Thyberg J, Terenius L, Nordstedt C. Controlling amyloid beta-peptide fibril formation with protease-stable ligands. J Biol Chem 1997; 272:12601 - 5; http://dx.doi.org/10.1074/jbc.272.19.12601; PMID: 9139713
- Findeis MA, Musso GM, Arico-Muendel CC, Benjamin HW, Hundal AM, Lee JJ, Chin J, Kelley M, Wakefield J, Hayward NJ, et al. Modified-peptide inhibitors of amyloid beta-peptide polymerization. Biochemistry 1999; 38:6791 - 800; http://dx.doi.org/10.1021/bi982824n; PMID: 10346900
- Chalifour RJ, McLaughlin RW, Lavoie L, Morissette C, Tremblay N, Boulé M, Sarazin P, Stéa D, Lacombe D, Tremblay P, et al. Stereoselective interactions of peptide inhibitors with the beta-amyloid peptide. J Biol Chem 2003; 278:34874 - 81; http://dx.doi.org/10.1074/jbc.M212694200; PMID: 12840031
- Bieschke J, Herbst M, Wiglenda T, Friedrich RP, Boeddrich A, Schiele F, Kleckers D, Lopez del Amo JM, Grüning BA, Wang Q, et al. Small-molecule conversion of toxic oligomers to nontoxic β-sheet-rich amyloid fibrils. Nat Chem Biol 2012; 8:93 - 101; http://dx.doi.org/10.1038/nchembio.719; PMID: 22101602
- Jiang L, Liu C, Leibly D, Landau M, Zhao M, Hughes MP, Eisenberg DS. Structure-based discovery of fiber-binding compounds that reduce the cytotoxicity of amyloid beta. Elife 2013; 2:e00857; http://dx.doi.org/10.7554/eLife.00857; PMID: 23878726
- Wiesehan K, Buder K, Linke RP, Patt S, Stoldt M, Unger E, Schmitt B, Bucci E, Willbold D. Selection of D-amino-acid peptides that bind to Alzheimer’s disease amyloid peptide abeta1-42 by mirror image phage display. Chembiochem 2003; 4:748 - 53; http://dx.doi.org/10.1002/cbic.200300631; PMID: 12898626
- Wiesehan K, Stöhr J, Nagel-Steger L, van Groen T, Riesner D, Willbold D. Inhibition of cytotoxicity and amyloid fibril formation by a D-amino acid peptide that specifically binds to Alzheimer’s disease amyloid peptide. Protein Eng Des Sel 2008; 21:241 - 6; http://dx.doi.org/10.1093/protein/gzm054; PMID: 18252750
- van Groen T, Wiesehan K, Funke SA, Kadish I, Nagel-Steger L, Willbold D. Reduction of Alzheimer’s disease amyloid plaque load in transgenic mice by D3, A D-enantiomeric peptide identified by mirror image phage display. ChemMedChem 2008; 3:1848 - 52; http://dx.doi.org/10.1002/cmdc.200800273; PMID: 19016284
- van Groen T, Kadish I, Wiesehan K, Funke SA, Willbold D. In vitro and in vivo staining characteristics of small, fluorescent, Abeta42-binding D-enantiomeric peptides in transgenic AD mouse models. ChemMedChem 2009; 4:276 - 82; http://dx.doi.org/10.1002/cmdc.200800289; PMID: 19072935
- Aileen Funke S, van Groen T, Kadish I, Bartnik D, Nagel-Steger L, Brener O, Sehl T, Batra-Safferling R, Moriscot C, Schoehn G, et al. Oral treatment with the d-enantiomeric peptide D3 improves the pathology and behavior of Alzheimer’s Disease transgenic mice. ACS Chem Neurosci 2010; 1:639 - 48; http://dx.doi.org/10.1021/cn100057j; PMID: 22778851
- Kokkoni N, Stott K, Amijee H, Mason JM, Doig AJ. N-Methylated peptide inhibitors of beta-amyloid aggregation and toxicity. Optimization of the inhibitor structure. Biochemistry 2006; 45:9906 - 18; http://dx.doi.org/10.1021/bi060837s; PMID: 16893191
- Amijee H, Bate C, Williams A, Virdee J, Jeggo R, Spanswick D, Scopes DI, Treherne JM, Mazzitelli S, Chawner R, et al. The N-methylated peptide SEN304 powerfully inhibits Aβ(1-42) toxicity by perturbing oligomer formation. Biochemistry 2012; 51:8338 - 52; http://dx.doi.org/10.1021/bi300415v; PMID: 23025847
- Matharu B, El-Agnaf O, Razvi A, Austen BM. Development of retro-inverso peptides as anti-aggregation drugs for β-amyloid in Alzheimer’s disease. Peptides 2010; 31:1866 - 72; http://dx.doi.org/10.1016/j.peptides.2010.06.033; PMID: 20633587
- Parthsarathy V, McClean PL, Hölscher C, Taylor M, Tinker C, Jones G, Kolosov O, Salvati E, Gregori M, Masserini M, et al. A novel retro-inverso peptide inhibitor reduces amyloid deposition, oxidation and inflammation and stimulates neurogenesis in the APPswe/PS1ΔE9 mouse model of Alzheimer’s disease. PLoS One 2013; 8:e54769; http://dx.doi.org/10.1371/journal.pone.0054769; PMID: 23382963
- Frydman-Marom A, Rechter M, Shefler I, Bram Y, Shalev DE, Gazit E. Cognitive-performance recovery of Alzheimer’s disease model mice by modulation of early soluble amyloidal assemblies. Angew Chem Int Ed Engl 2009; 48:1981 - 6; http://dx.doi.org/10.1002/anie.200802123; PMID: 19035593
- Holmes C, Boche D, Wilkinson D, Yadegarfar G, Hopkins V, Bayer A, Jones RW, Bullock R, Love S, Neal JW, et al. Long-term effects of Abeta42 immunisation in Alzheimer’s disease: follow-up of a randomised, placebo-controlled phase I trial. Lancet 2008; 372:216 - 23; http://dx.doi.org/10.1016/S0140-6736(08)61075-2; PMID: 18640458
- Sperling RA, Aisen PS, Beckett LA, Bennett DA, Craft S, Fagan AM, Iwatsubo T, Jack CR Jr., Kaye J, Montine TJ, et al. Toward defining the preclinical stages of Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement 2011; 7:280 - 92; http://dx.doi.org/10.1016/j.jalz.2011.03.003; PMID: 21514248
- Crescenzi O, Tomaselli S, Guerrini R, Salvadori S, D’Ursi AM, Temussi PA, Picone D. Solution structure of the Alzheimer amyloid beta-peptide (1-42) in an apolar microenvironment. Similarity with a virus fusion domain. Eur J Biochem 2002; 269:5642 - 8; http://dx.doi.org/10.1046/j.1432-1033.2002.03271.x; PMID: 12423364