
Abstract
Prion diseases are infectious and inevitably fatal neurodegenerative diseases characterized by prion replication, widespread protein aggregation and spongiform degeneration of major brain regions controlling motor function. Oxidative stress has been implicated in prion-related neuronal degeneration, but the molecular mechanisms underlying prion-induced oxidative damage are not well understood. In this study, we evaluated the role of oxidative stress-sensitive, pro-apoptotic protein kinase Cδ (PKCδ) in prion-induced neuronal cell death using cerebellar organotypic slice cultures (COSC) and mouse models of prion diseases. We found a significant upregulation of PKCδ in RML scrapie-infected COSC, as evidenced by increased levels of both PKCδ protein and its mRNA. We also found an enhanced regulatory phosphorylation of PKCδ at its two regulatory sites, Thr505 in the activation loop and Tyr311 at the caspase-3 cleavage site. The prion infection also induced proteolytic activation of PKCδ in our COSC model. Immunohistochemical analysis of scrapie-infected COSC revealed loss of PKCδ positive Purkinje cells and enhanced astrocyte proliferation. Further examination of PKCδ signaling in the RML scrapie adopted in vivo mouse model showed increased proteolytic cleavage and Tyr 311 phosphorylation of the kinase. Notably, we observed a delayed onset of scrapie-induced motor symptoms in PKCδ knockout (PKCδ−/−) mice as compared with wild-type (PKCδ+/+) mice, further substantiating the role of PKCδ in prion disease. Collectively, these data suggest that PKCδ signaling likely plays a role in the neurodegenerative processes associated with prion diseases.
Introduction
Prion diseases, or transmissible spongiform encephalopathies (TSEs), are a group of fatal neurodegenerative diseases of humans and animals. Human prion diseases include Creutzfeldt-Jakob disease (CJD), Kuru, Gerstmann-Sträussler-Scheinker disease (GSS) and Fatal familial insomnia (FFI). Examples of TSEs found in animals include bovine spongiform encephalopathy (BSE), scrapie in sheep, chronic wasting disease (CWD) in deer and elk, and transmissible mink encephalopathy (TME).Citation1-Citation3 All these diseases share common neuropathological and clinical signs. Prion diseases are mainly characterized by misfolding of the normal cellular prion protein (PrPC) into the pathological isoform PrPSc through unknown pathogenic mechanisms. Abnormally folded PrPSc is insoluble in non-denaturing detergents and partially resistant to protease digestion. Prion diseases generally involve long incubation periods, characteristic spongiform changes associated with protein aggregation and neuronal loss, and widespread gliosis.
PrPC is a glycosylphosphatidylinositol (GPI)-linked extracellular membrane protein, which is ubiquitously expressed in the central nervous system, in both neuronal and glial cells.Citation1 The biological function of PrPC is poorly understood, but evidence suggests that this protein functions as an antioxidant and metal-binding protein with a role in several cellular processes in the nervous system, including neurite outgrowth, synapse formation, and maintenance of myelinated axons.Citation1,Citation2,Citation4-Citation6 During disease progression, abnormal conformational changes in PrPC result in pathogenic PrPSc molecules, yet the mechanism by which this change occurs is unknown.Citation7 Studies have shown that propagation of the infectious prion protein cannot occur in the absence of host prion proteins,Citation8 indicating a causal role for prion proteins in pathogenesis.
Prion diseases cause severe neuronal damage mainly in brain regions that coordinate motor function, including the basal ganglia, cerebral cortex, thalamus, and cerebellum. Current knowledge about the molecular mechanisms underlying neurodegeneration in prion disease and related prionopathies is limited. However, growing evidence indicates that oxidative stress and apoptosis contribute to the neurodegenerative process in prion disease pathogenesis.Citation9-Citation11 Therefore, furthering our understanding of molecular signaling mechanisms involving early pathologic changes in prion disease is important for developing intervention strategies. We have previously shown that protein kinase Cδ (PKCδ) is an oxidative stress-sensitive, pro-apoptotic kinase, which plays a causal role in apoptotic neuronal cell death.Citation12-Citation15 We demonstrated that phosphorylation of tyrosine residue 311 and caspase-3-dependent proteolytic cleavage result in a persistent increase in the kinase activity, which promotes neuronal apoptosis.Citation13 Herein, we evaluate the role of PKCδ in prion neurotoxicity in ex vivo brain slice culture and in vivo experimental models of infectious prion disease. Our results reveal that PKCδ proteolytic signaling plays an important role in the neuronal damage and neurological deficits of prion disease. Thus, our findings provide further mechanistic insight into pathogenesis of prion diseases.
Results
Upregulation of PKCδ in RML-infected cerebellar organotypic slice culture model of prion disease
Brain slice models preserve the tissue architecture and neural connectivity of the brain regions, making them ideal platforms to model the neuropathology of brain atrophy.Citation16,Citation17 Cerebellar organotypic slice cultures (COSC) have recently been shown to retain and replicate prion infection and thereby serve as an excellent ex vivo model to study the pathogenesis of prion disease.Citation18,Citation19 Evidence has indicated that prion titers in COSC peaked at 4 week, while at 5 week of infection, the PrPSc titers correspond to terminal infection in mice 5 month post-infection.Citation19 In this study, we first investigated whether COSCs from wild-type C57BL/6 mice reproduce pathological changes of prion diseases. Since gliosis is a well-established integral part of prion infections, we evaluated the glial cell proliferation following RML infection by immunocytochemistry after two weeks of infection. Astrogliosis was enhanced in RML-infected COSCs, as evidenced by an enhanced GFAP immunofluorescence signal (), indicating that the slice cultures undergo the neurotoxic stress at the early stages of prion infection. Next, we examined whether PKCδ expression was altered during two weeks of prion infection by immunocytochemical analysis of PKCδ. As shown in , PKCδ immunoreactivity in RML-infected COSCs was significantly higher than in the slices inoculated with normal brain homogenate (NBH). These data suggest that PKCδ upregulation may have a role in prion pathogenesis.
Figure 1. Prion replication and PKCδ upregulation in RML scrapie-infected organotypic cerebellar slice culture. (A) Immunohistochemical analysis of widespread astrogliosis and PKCδ upregulation in RML scrapie-infected cerebellar slice culture (right panel) as compared with control NBH-inoculated cerebellar slice culture (left panel). (B) Western blot analysis of the proteinase-K resistant PrPSc isoform after five weeks of RML infection in cerebellar slice cultures.
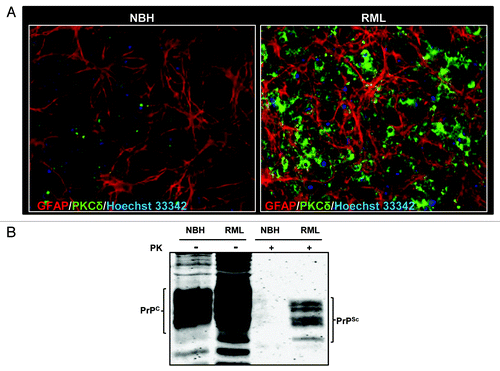
We also evaluated whether COSCs retain PrPsc infection for five weeks following RML brain homogenate inoculation. For this purpose, RML-infected or NBH-inoculated slices were subjected to limited proteolysis. This process completely digests PrPC, whereas the C-terminal region of truncated PrPSc remains intact (these fragments are referred to as PrPcore), which is a disease-specific marker of prion infection.Citation20 Following PK digestion, protein lysates were separated by western blot and membrane probed with the prion antibody POM-1, which specifically recognizes amino acid 121–230 at the globular domain of PrP.Citation21,Citation22 As shown in , after PK digestion, characteristic di-, mono- and unglycosylated forms of PK-resistant bands were observed only in RML scrapie-exposed slices, while they were completely digested in slices exposed to uninfected NBH, confirming that COSCs retain the prion infection over five weeks.
PKCδ activation during RML infection in organotypic slice cultures
We have previously shown that proteolytic activation of PKCδ plays an important role in the oxidative stress-induced apoptotic cell death in dopaminergic neurons.Citation12,Citation23-Citation25 It has also been reported that oxidative stress,Citation26,Citation27 mitochondrial dysfunction,Citation28,Citation29 and endoplasmic reticular stressCitation4 are involved in the pathogenesis of neurodegenerative diseases, including prion diseases. In this study, we evaluated PKCδ activation in the early stages of RML infection in COSC exposed to NBH or RML for 14 d. Western blot analysis using the antibody that recognizes both native (72–74kDa) and cleaved PKCδ proteins (38–41kDa) showed a significant upregulation of native PKCδ as well as cleaved PKCδ protein fragments in RML-infected cultures (). We previously demonstrated that phosphorylation of tyrosine 311 is required for caspase-3-mediated proteolytic activation of PKCδ 13 and its pro-apoptotic function.Citation4,Citation15,Citation23,Citation30,Citation31 In addition to tyrosine 311 phosphorylation, PKCδ-Thr505 activation-loop phosphorylation is required for full kinase activity of PKCδ, and thus serves as a marker of PKCδ activation.Citation32 Therefore, we determined the phosphorylation of Tyr311 and Thr505 in prion-infected COSCs by western blot using phospho-specific antibodies. As shown in and , RML infection significantly increased Tyr311 and Thr505 phosphorylation compared with control NBH-inoculated slice cultures.
Figure 2. Proteolytic activation of PKCδ in RML scrapie-infected organotypic cerebellar slice culture. (A) Representative western blots for PKCδ upregulation, cleavage and phosphorylation upon RML-scrapie infection in cerebellar slice cultures. Densitometric analysis of western blots for (B) Native PKCδ (C) Cleaved PKCδ (D) p-PKCδ (Thr505) and (E) p-PKCδ (Tyr311) upregulation upon RML scrapie infection. Each group represented as mean ± SEM from at least five separate measurements. (F) qRT-PCR analysis for PKCδ mRNA expression following RML scrapie infection. Each group represented as mean ± SEM from at least six measurements from three separate experiments. (G) PKCδ immunoprecipitation (IP)-kinase assay for augmented kinase activity upon RML scrapie infection in organotypic slice cultures. Each group represented as mean ± SEM from at least four measurements from two separate experiments (*P < 0.05 vs NBH, **P < 0.01 vs NBH, ***P < 0.001 vs NBH).
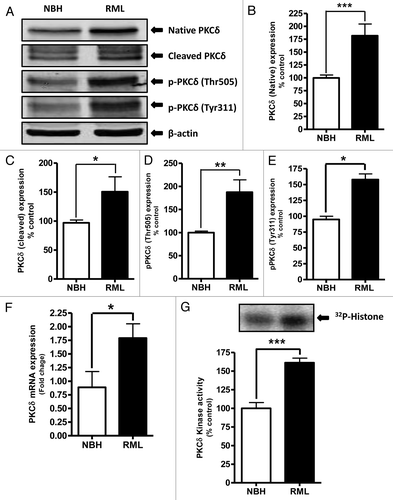
Next, we employed the Citation32p IP-kinase assay to determine if RML-induced proteolytic activation of PKCδ is associated with a sustained activation of its kinase activity in organotypic slice cultures. PKCδ kinase assays performed in the absence of lipid cofactors showed a robust increase in PKCδ kinase activity in RML-infected samples compared with NBH-inoculated samples (), indicating that RML-induced proteolytic activation of PKCδ is associated with increased kinase activity. To further confirm the PKCδ upregulation, we quantified PKCδ mRNA levels upon exposure to either NBH or RML scrapie infection by quantitative-RT-PCR analysis. Interestingly, our results show that RML infection leads to increased levels of PKCδ mRNA (), suggesting the RML infection-meditated upregulation of PKCδ is through transcriptional induction. Collectively, our results in the COSC model demonstrate that prion infection activates PKCδ kinase activity by proteolytic cleavage and upregulation of the kinase.
Cerebellar atrophy during RML infection
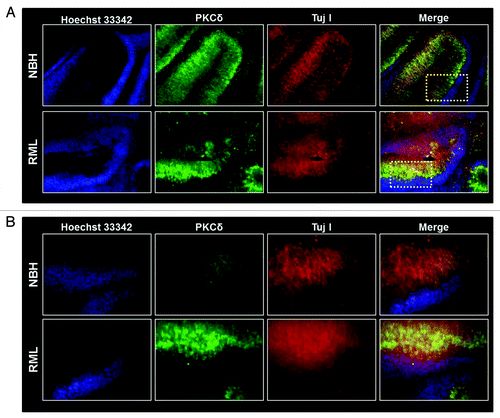
To evaluate the effect of RML infection on the cerebellum, we infected organotypic cerebellar cultures with either NBH or RML for two weeks, as described above. At the end of the incubation, cerebellar slices were analyzed for cerebellar atrophy induced by prion infection. As shown in , compared with slices inoculated with NBH, cultures infected by RML have more diffuse staining for TujI, a neuronal marker for neuron-specific class III β-tubulin, indicating that RML infection induces discernible cerebellar degeneration. Additionally, immunostaining for PKCδ indicates an intense PKCδ immunoreactivity in the RML-infected cerebellar brain slices, predominantly in the Purkinje cells lining the molecular layer. Histological evaluation of the RML-infected COSCs indicates a marked cerebellar atrophy compared with NBH-inoculated slices. In RML-infected cerebella slices, extensive degeneration was observed in the molecular layer, similar to previous reports evaluating neuropathological changes in human CJD and GSS incidents.Citation33
Figure 3. Cerebellar degeneration during RML infection in organotypic cerebellar slice culture. (A) Immunohistochemical analysis of pronounced cerebellar atrophy during RML infection (lower panel) and intact healthy histological morphology in control NHB-inoculated (upper panel) cerebellar slices. White dotted boxes represent regions magnified in slices described below. (B) Representative high magnification (20X) images indicating possible neuronal damage (as seen by diffuse Tuj1 staining) and PKCδ upregulation during RML infection (lower panel).
In vivo proteolytic activation of PKCδ in murine-adapted RML scrapie-infected mice
Apoptosis has been characterized as a major mode of neuronal cell death associated with spongiform degeneration,Citation34,Citation35 and PKCδ has been identified as a key pro-apoptotic molecule involved in neuronal apoptosis.Citation12 To evaluate whether PKCδ activation occurs in vivo during TSE-induced neurodegeneration, we examined RML scrapie- and mock NBH-inoculated wild-type mice at 60, 90, 120 and 150 DPI for changes in native and proteolytically cleaved PKCδ fragments in cerebellar tissues. As shown in , increased levels of cleaved PKCδ in RML-infected animals were observed at preclinical stages (90 and 120 DPI) of TSE, compared with mock-infected animals. Quantified immunoblots for the cleaved PKCδ protein fragment () show a 75–80% induction of proteolytic activation in RML-infected animals beyond that expressed in NBH-inoculated animals at these preclinical stages, suggesting the induction of apoptotic stimuli leading to cerebellar atrophy-related behavioral deficits. However, we did not find increased native PKCδ protein levels, as observed in the RML-infected slice cultures ( and ). Furthermore, PKCδ protein level was slightly reduced at the time point (150 DPI) marking the terminal stages of TSE.
Figure 4. PKCδ activation in RML scrapie-infected mice. (A) Representative western blots for native, cleaved and phosphorylated PKCδ at 60, 90, 120 and 150 d post-infection (DPI). Densitometric analysis of western blots for changes in (B) Native PKCδ (C) Cleaved PKCδ (D) p-PKCδ (Tyr311) and (E) p-PKCδ (Thr505) upon RML infection at various time points. All data were normalized to NBH expression levels and are represented as mean percentage (± S.E.M.) of uninfected protein expression from at least three separate animals.
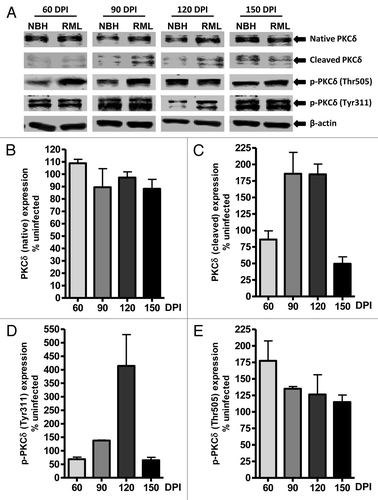
Next, we examined the phosphorylation of PKCδ at two regulatory sites: Tyr311 flanking the caspase-3 cleavage site of PKCδ and Thr505 within the activation loop. Analysis of Tyr311 phosphorylation upon RML infection ( and ) indicates a slight reduction at 60 DPI and then a significant increase in the phosphorylation signal at 90 and 120 DPI. It is noteworthy that the time-course pattern for this PKCδ cleavage-specific phosphorylation event is also reflected in the increase in cleaved PKCδ fragments at 90 and 120 DPI. This positive correlation indicates the potential significance of these events at these preclinical TSE stages. Additional quantitation of immunoblots for phosphorylated PKCδ at Thr505 () indicated about a 75% increase at 60 DPI and about a 25% increase of phosphorylation events at 90 and 120 DPI when normalized to Thr505 phosphorylation signals in NBH-inoculated samples at the respective time points. Together, our results in an animal model of prion disease revealed time-dependent alterations in PKCδ signaling.
TSE-related behavioral abnormalities are delayed in PKCδ (−/−) mice
The neurological symptoms of mice infected with mouse-adapted scrapie include kyphosis, tremor, cachexia, and ataxia.Citation36-Citation39 Additionally, infected mice display clasping of limbs when held aloft by the tail.Citation40 If PKCδ signaling contributes significantly to the neurodegeneration and neurological deficits associated with TSE, inhibition of PKCδ function would delay the onset of the disease. To further validate the role of PKCδ signaling in TSE, we utilized PKCδ knockout PKCδ (−/−) C57 black mice, as described in our previous publication.Citation41 Specifically, we evaluated the motor function in PKCδ (−/−) C57 black mice following inoculation with murine-adapted RML scrapie and compared with that of wild-type C57 black mice. After inoculation with either NBH or RML, mice were subjected to weekly behavioral analysis using the horizontal bar test and grip strength meter to assess grip strength, stamina, and coordination.
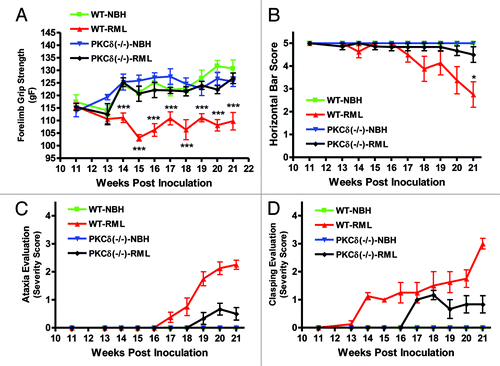
Beginning at week 14, wild-type RML-infected animals displayed significantly reduced forelimb strength, which continued until the end of the study (). Conversely, RML-infected PKCδ (−/−) animals retained forelimb strength until sacrifice at 150 DPI. For the horizontal bar test (), mice were scored from 0 to 5, reflecting increasing hang durations while suspended from the metal bar. Infected wild-type animals began showing difficulty hanging on the bar at week 18, and the score significantly decreased at week 21 (). PKCδ (−/−) mice showed no significant reduction during the course of infection. In addition to grip strength and horizontal bar measurements, general clinical evaluation of all mice was done weekly in order to identify behavioral signs of RML scrapie-induced motor deficits. We evaluated both wild-type and PKCδ (−/−) mice for differences in motor signs that reflect difficulty initiating ambulatory activities and changes in ambulatory activity patterns. We scored their pathology on a scale from 0 to 4, with 0 indicating normal movement and 4 indicating almost complete lethargy and ataxia (). Test animals were monitored over a period of several minutes for changes in behavioral motor deficits reflecting postural instability and difficulty in open field ambulation. Symptoms began appearing in wild-type mice at week 17 and became progressively more pronounced over the course of monitoring. By contrast, PKCδ (−/−) mice showed a delayed onset of TSE-related ataxia, and their observable motor signs were scored as less severe. Additionally, mice were tested for the clasping of limbs when held aloft by the tail. PKCδ (−/−) mice likewise showed a delay in the onset of clasping, as well as reduced severity scores (). Taken together, these results indicate that PKCδ knockout delays the onset of neurological signs associated with mouse-adapted scrapie in animal models.
Figure 5. PKCδ knockout mice are resistant to RML scrapie-induced motor deficits. RML scrapie-infected WT and PKCδ (−/−) mice were evaluated weekly for motor changes after inoculation. A) Forelimb grip strength was significantly reduced beginning at 14 wk in wild-type mice, whereas PKC (−/−) animals did not differ significantly from mock-infected wild-type animals throughout the course of infection. B) Motor function was evaluated using the horizontal bar test. Wild-type RML scrapie-infected mice began to show deficits at 17 wk, and changes became significant at 21 wk. RML scrapie-infected PKCδ (−/−) mice and mock-infected mice showed no significant changes. C) Mice were evaluated for open-field ambulation and their ability to initiate movement over the course of several minutes. Slight changes in motor function were observed in infected WT mice beginning at 18 wk, progressing to significant ataxia by 21 wk. Conversely, PKCδ (−/−) mice displayed a delayed onset in motor signs, and changes remained mild throughout the monitoring period. D) The clasping of limbs when held aloft by the tail was evaluated in WT and PKCδ (−/−) mice. Clasping symptoms began in WT mice at 14 wk and steadily progressed to severe over the course of monitoring. PKCδ (−/−) animals did not begin clasping limbs until 17 wk, and symptoms were mild over the course of monitoring. Data represented as mean ± SEM for each group.
Discussion
In this study, we report that PKCδ is proteolytically activated during the preclinical stages of TSE in our experimental models of prion disease. Our results also demonstrate that phosphorylation of PKCδ at its activation loop and its catalytic domain is important for disease progression and neuronal cell death during the course of prion infection. To the best of our knowledge, we are the first to demonstrate that PKCδ (−/−) mice exhibit delayed onset of the behavioral symptoms associated with the prion disease. These findings could contribute to the development of interventional strategies for TSE-related motor deficits.
One of the main limitations for characterizing the molecular mechanisms of TSE is the lack of suitable in vitro experimental models of infectious prion disease. Indeed, several in vitro experimental models, such as neuronal cell lines infected with mouse-adapted scrapie, have been used to investigate the biochemical properties of PrPSc with some success at expanding the horizons of prion biology. However, due to several limitations associated with cell culture systems, they are not amenable for studying in-depth molecular mechanisms underlying infectious prion disease. Recently, Falsig and his colleagues successfully developed and validatedCitation18,Citation19,Citation42 an ex vivo transmission model for prion disease using organotypic cerebellar slice cultures. In this study, we successfully adopted this organotypic cerebellar slice culture to study PKCδ-mediated neuronal cell death during TSE. First, we were able to reproduce the RML-infected organotypic cerebellar slice cultures in our laboratory with slight modifications. We observed an improved viability of cerebellar slices with use of Compresstome™ Vf-300 microtome, because this procedure significantly reduced the cutting time and mechanical stress associated with slice preparation. Our slice cultures were able to retain infection, as seen by widespread gliosis during early stages of the infection (2 wk, ) and by the presence of PK-resistant prion proteins after 5 wk of RML incubation (). Following establishment of COSCs with prion infection, we systematically studied the role of proteolytically activated pro-apoptotic kinase PKCδ in prion-induced neurodegeneration by comparing the kinase signaling in RML-infected and mock NBH-inoculated slice cultures.
We have previously shown that PKCδ is a key oxidative stress-sensitive kinase that can be activated by caspase-3-dependent proteolytic cleavage and by tyrosine and threonine phosphorylation.Citation23,Citation24,Citation43,Citation44 Also, oxidative stress has been identified as integral to prion-induced neurodegeneration.Citation9-Citation11,Citation45,Citation46Therefore, herein, we have evaluated PKCδ-mediated neuronal apoptosis in ex vivo cerebellar slice cultures and in RML-infected in vivo mice. Our data support the idea of PKCδ proteolytic activation during prion infection, as noted by the significantly increased native and cleaved PKCδ upon RML infection in COSCs. We also observed increased phosphorylation of PKCδ Tyr311 and Thr505 levels in RML-infected COSCs, compared with mock-infected tissues. Phosphorylation of Tyr311 at the hinge region of PKCδ, which reportedly causes conformational changes in the structure that opens the catalytic domain,Citation47 mediates caspase-3-dependent cleavage, thereby separating the catalytic and regulatory domains.Citation13,Citation25 This proteolytic activation of PKCδ promotes apoptosis by activating downstream apoptotic cascades.Citation43,Citation48 In addition, the cleaved catalytically active PKCδ fragment activates upstream caspase signaling, constituting a positive feedback mechanism leading to a further amplified apoptotic pathway.Citation48-Citation50 Furthermore, analysis of the PKCδ activation loop-specific site Thr505 indicates that RML infection could induce phosphorylation of Thr505 to further enhance the kinase activation. These observations were further validated by the upregulation of PKCδ kinase activity, as seen by PKCδ IP-kinase assays. We also analyzed the PKCδ mRNA and the native protein levels upon RML infection. Surprisingly, we observed increased PKCδ mRNA expression and protein levels in infected slices, indicating transcriptional upregulation of native PKCδ expression. Next, we performed immunohistochemical analysis of the extent of neurodegeneration in RML-infected cerebellar slices. We observed widespread neuronal cell death (), as seen by diffused staining for the neuronal marker Tuj1 and increased PKCδ staining in RML-infected tissues compared with mock NBH-inoculated tissues. Also, we morphologically discerned cerebellar atrophy in the RML-infected slices, which can be extrapolated to cerebellar dysfunction and locomotor deficits involved with TSE.
Following the evaluation of PKCδ in organotypic slice cultures, we further characterized the role of PKCδ in in vivo experimental models of prion disease by intracerebrally inoculating mouse-adapted RML scrapie or NBH to wild-type C57BL/6 mice and analyzing PKCδ signals 60, 90, 120 and 150 d post-infection. Biochemical analysis of cerebellar tissues in these mice revealed significantly increased levels of cleaved PKCδ protein during preclinical stages, indicating the possible involvement of neuronal cell death. These findings corroborate with previous reports, indicating that oxidative stress is induced during early prion invasion, thus predisposing the brain to neurodegeneration.Citation51 We also observed increased phosphorylation of PKCδ Tyr311 levels, via immunoblotting, which in combination with the proteolytic activation of PKCδ, makes neurons susceptible to downstream apoptotic cascades. Since PKCδ is abundantly expressed in the cerebellum,Citation52 especially in Purkinje cells and the posterior cerebellar cortex,Citation53 these neurons may be more susceptible to TSE-induced neurotoxic stress and more likely to undergo degeneration resulting from the disease. Increased PKCδ cleavage without any significant change in the native kinase at 90 and 120 DPI suggests that PKCδ upregulation may have been compensated by increased proteolysis of the kinase at the early stages of the infection. The observed decrease in native PKCδ as well as the cleaved fragment at the terminal stage of the disease may be due to the loss of neurons normally expressing higher levels of PKCδ.
Since TSE is a fatal neurodegenerative disease often involving motor and postural abnormalities associated with cerebellar dysfunction,Citation39,Citation54,Citation55 we performed several behavioral tests in RML-infected mice throughout the study. For our behavior testing, we included the transgenic PKCδ (−/−) mice to evaluate whether abolition of PKCδ would alter TSE-associated neurobehavioral deficits. We evaluated ataxia, hind limb clasping phenotype, forelimb grip strength and horizontal bar performance to assess neurological damage, particularly cerebellar ataxia and other motor deficits.Citation56-Citation58 For most of the behavioral tests, wild-type animals infected with RML started showing behavioral deficits around 14–17 wk post-infection, whereas mice receiving the mock NBH inoculation did not show any behavior deficits throughout the study. Interestingly, PKCδ (−/−) showed a delayed onset of the motor deficits associated with TSE, suggesting that PKCδ signaling plays as a major role in the pathogenesis of prion disease. We have previously shown that the PKCδ inhibitor rottlerin effectively protected against locomotor deficits in an animal model of dopaminergic neurodegeneration.Citation41 Therefore, successful attenuation of scrapie-induced neurodegeneration by pharmacological inhibition of PKCδ could provide a basis for developing therapies effective prior to the onset of widespread spongiform neurodegeneration and motor deficits in TSE.
In conclusion, we have demonstrated an enhanced regulatory phosphorylation and proteolytic activation of PKCδ during the progression of mouse-adapted scrapie cerebellar slice cultures and a delayed onset of scrapie-induced motor symptoms in PKCδ knockout mice, suggesting a possible role of PKCδ in the neurotoxicity of prion disease. These findings may help to further elucidate the PKCδ-dependent cell signaling mechanism in TSE pathogenesis and to develop potential pharmacological interventions for TSE.
Materials and Methods
Prion organotypic slice culture assay (POSCA)
Iowa State University’s (ISU) College of Veterinary Medicine is accredited by the Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC), and all procedures involving animal handling were approved by the Institutional Animal Care and Use Committee (IACUC) at ISU. Organotypic slice cultures were prepared as previously described with some modifications.Citation19,Citation42 Briefly, brain blocks were prepared in 2% (w/v) low-melting-point agarose (Invitrogen 15517–022), and 350-µm thick cerebellar slices were prepared from 9–12 d-old C57BL/6 pups using a Compresstome™ Vf-300 microtome (Precisionary Instruments Inc.). Slices were inoculated with 100 µg of 1% normal brain homogenate (NBH) or Rocky Mountain Laboratory (RML)-infected brain homogenate per 10 slices in Gey’s balanced salt solution (GBSS: 137 mM NaCl, 5 mM KCl, 0.845 mM Na2HPO4, 1.5 mM CaCl2.2H2O, 0.66 mM KH2PO4, 0.28 mM MgSO4.7H2O, 1.0 mM MgCl2.6H20, 2.7 mM NaHCO3 and pH-adjusted to 7.4) supplemented with 1 mM of the glutamate receptor antagonist kynurenic acid. Free-floating slices were incubated for 1 h in the brain homogenate at 4 °C, washed twice and transferred to Millicell-CM Biopore PTFE membrane inserts (Millipore PICM03050) and maintained in slice culture medium (50% MEM, 25% Basal Eagle medium, 25% horse serum, 0.65% glucose supplemented with penicillin/streptomycin and glutamax). Organotypic slice cultures were maintained in a humidified 37 °C incubator with 5% CO2 and 95% air for 14 d, and 90% of the media was exchanged every other day.
Immunohistochemistry
For immunohistochemistry, the organotypic slices were washed with PBS and fixed with 4% paraformaldehyde (PFA) for 1–2 h at room temperature. After fixing, the membrane inserts containing cerebellar slice cultures were washed with PBS and incubated with the blocking agent (2% goat serum, and 0.1% Triton X-100 in PBS) for 1 h. Membrane inserts were then incubated with the following combinations of primary antibodies: anti-GFAP (1:1000, Millipore) and anti-PKCδ (1:1000, Santa Cruz), or anti-β III tubulin (Tuj1) (1:1000, Millipore) and anti-PKCδ (1:1000, Santa Cruz) for 2–3 d at 4 °C. At the end of the incubation, membranes were washed with PBS and incubated with Alexa Fluor 555-conjugated anti-mouse secondary antibody (1:2000) or Alexa Fluor 488-conjugated anti-rabbit secondary antibody (1:2000) for 90 min in the dark. Hoechst 44432 was used as a nuclear stain. The culture membranes were removed from the inserts and mounted directly on microscope slides using Fluoromount mounting medium (Sigma) and viewed using a Nikon TE2000 microscope (Tokyo, Japan). Images were captured with a SPOT color digital camera (Diagnostic Instruments, Sterling Heights, MI).
Western blot
Slice cultures were washed twice in ice-cold PBS and lysed with Tissue extraction reagent (Invitrogen FNN0071) containing protease and phosphatase inhibitor cocktail (Thermo Scientific). Whole tissue lysates were prepared as described.Citation44 Protein concentrations were determined with the Bradford protein assay kit (Bio-Rad). Immunoblot analysis was performed as previously described.Citation59,Citation60 Briefly, the indicated protein lysates containing equal amounts of protein were separated on a 12–15% SDS-polyacrylamide gel. After separation, proteins were transferred to a nitrocellulose membrane, and nonspecific binding sites were blocked by treating with LI-COR blocking buffer. The membranes were then incubated overnight with primary antibody directed against PKCδ (1:1000, Santa Cruz), pPKCδ-Thr505 (1:1000, Santa Cruz), pPKCδ-Tyr311 (1:1000, Santa Cruz), or PrP (POM-1, 1:5000, Prionatis AG). The primary antibody treatments were followed by treatment with IR800-conjugated anti-rabbit or Alexa Fluor 680-conjugated anti-mouse secondary antibody for 1 h at room temperature. To confirm equal protein loadings, blots were reprobed with β-actin antibody (1:15000 dilutions). Western blot images were captured with the Odyssey IR Imaging system (LI-COR) and data were analyzed using Odyssey 2.0 software.
Limited proteolysis of prion protein resistance to proteinase-K (PK) digestion was measured as previously described with few modifications.Citation19,Citation42 RML-infected and NBH-inoculated slice cultures were homogenized 35 d after infection in lysis buffer (50 mM TRIS-HCl, pH 7.4, 0.5% Triton X, and 0.5% sodium deoxycholate). Protein concentration was determined by Bradford assay. Next, 40 µg of protein was digested with 25 μg/ml of proteinase-K and incubated for 30 min. The reaction was stopped by boiling the samples at 95 °C for 7 min prior to analysis by western blot as described above.
qRT-PCR
Organotypic slice cultures were washed once with PBS, and total RNA was isolated using the Absolutely RNA Miniprep kit from Agilent. Next, 500 ng of total RNA was converted to cDNA using the High Capacity cDNA Archive kit (Applied Biosystems). Quantitative real-time RT-PCR was performed using the Brilliant SYBR Green qPCR Master Mix kit (Qiagen) in an Mx3000P qPCR system (Agilent), as described previously.Citation23 The PKCδ primer set (Qiagen; QT00107513) was used to analyze relative gene expression with the 18s primer set (Qiagen; QT02448075) as the internal control for RNA quantity. The PCR reaction mixture included 2 µl of cDNA, 10 µl of 2× master mix, and 0.2 µM of each primer. A negative control lacking cDNA (no template control) was included in each assay. The PCR cycle conditions were as follows: 95 °C for 10 min, then 40 cycles of 95 °C for 15 s and 60 °C for 1 min. The cycle threshold (Ct) values corresponding to the PCR cycle number at which fluorescence emission in real-time reaches a threshold above the baseline emission were determined. The relative PKCδ expression was calculated after adjusting for 18s using 2-ΔΔCt, where ΔCt is the PKCδ gene Ct -18s Ct.
PKCδ immunoprecipitation (IP)-kinase assays
The PKCδ enzymatic kinase activity assay was performed as described previously.Citation30 Briefly, RML scrapie- or NBH-infected slices were lysed with the lysis buffer (25 mM HEPES, 20 mM β-Glycerophosphate, 0.1 mM Na3VO4, 0.1% Triton-X, 300 mM NaCl, 1.5 mM MgCl2, 0.2 mM EDTA, 0.5 mM DTT and 10 mM NaF) containing protease and phosphatase inhibitor cocktail (Thermo Scientific). The lysates were placed on ice for 20 min, sonicated gently and centrifuged at 12 000 g for 45 min. After collecting the supernatant, protein concentration was determined using the Bradford assay. Next, 500μg of total protein in a 250-μl volume was immunoprecipitated overnight at 4 °C using 10μg of the PKCδ antibody. After adding protein A-agarose beads (Sigma-Aldrich) the next day, the samples were incubated for 2h at room temperature. The protein A-bound antibody complexes were then washed three times in 2× kinase assay buffer (40mM Tris, pH 7.4, 20mM MgCl2, 20μM ATP, and 2.5mM CaCl2), and then resuspended in 100μl of the same buffer. The kinase reaction was started by adding 100μl of the reaction buffer containing 0.4mg of histone H1, 50μg/ml phosphatidylserine, 4μM dioleoylglycerol, and 10 μCi of [γ-32P] ATP at 3000Ci/mM to the immunoprecipitated samples. The samples were incubated for 10 min at 30 °C. The kinase reaction was stopped by adding 2 × SDS-loading buffer and boiling the samples for 5 min. The proteins were separated on a 15% SDS-PAGE gel and the phosphorylated histone H1 bands were scanned using a Fujifilm FLA 5000 imager. Image analysis and band quantification were performed using the Fujifilm Multigauge software package (Fujifilm USA).
Mouse models of murine scrapie
Mouse models are a popular and versatile in vivo method for studying the effects of prion proteins and for providing insights into the neurodegenerative mechanisms of prion disease. All work with prion-infected strains and animals was performed under the licensure granted by a United States Department of Agriculture (USDA) permit for importation and exportation of controlled materials and it conformed to guidelines of the IACUC at ISU. Wild type PKCδ (+/+) and PKCδ (−/−) C57 black mice at postnatal week 6–8 were arranged by weight and randomized into RML and mock NBH inoculation groups. PKCδ knockout animals were originally obtained from Dr. Keiichi Nakayama at the Medical Institute of Bioregulation, Fukuoka, Japan.Citation61 RML and NBH infections were performed by intracerebrally inoculating 30 μl of 1% w/v RML brain homogenate into deeply anesthetized mice using a 27 gauge needle. Mice were monitored for 48 h post-inoculation for adverse effects and euthanized if they displayed unequivocal neurological signs. At 60, 90, 120, and 150 d post inoculation (DPI), all animals were subjected to behavioral evaluations described below, then following euthanasia, brain tissues were extracted for biochemical evaluations.
Behavioral evaluations
Extensive neuronal loss during the course of transmissible spongiform encephalopathy (TSE) produces progressive motor deficits in humans and animals including ataxia, tremor, and postural instability. Grip stamina and motor control were evaluated using the horizontal bar test.Citation62,Citation63 Each mouse was held by the tail and allowed to grip a 0.2 cm diameter brass rod 50 cm above a padded cage with its forelimbs. The padded Innovive mouse cage was placed below the bar to protect the mice from injury by falling. The mouse was quickly released and the time it took for the mouse to fall was recorded. Scores were given based on the following paradigm: 0–5s = 1; 6–10s = 2; 11–20s = 3; 21–29s = 4; and 30s or reaching the side support = 5.
The Columbus Instrument grip strength meter was used to determine the maximal amount of force that each animal applied with its forelimbs to a specially designed bar. For all measurements, the gauge was measured in PEAK mode and units were set to kgF. Animals were gripped firmly but gently at the base of the tail and the animal’s forelimbs were placed on the grip bar. Once the animal gripped the bar with both forelimbs, it was steadily pulled directly away from the digital strength meter. This process was repeated 2–3 times to get the peak strength reading.
Behavioral evaluations were done by trained personnel in cooperation with ISU Laboratory Animal Resources. We evaluated uninfected and RML scrapie-infected C57BL/6 and PKCδ (−/−) mice once-weekly for differences in motor signs that reflected difficulty initiating ambulatory activities and changes in ambulatory activity patterns. Motor performance was scored on a scale from 0 to 4 with 0 = normal movement, 1 = slight alteration in ambulation, 2 = obvious intermittent motor signs, 3 = continuous pronounced motor signs, and 4 = almost complete lethargy. Test animals were monitored for changes in behavioral motor deficits over a period of several minutes. Mice were also evaluated for clasping of limbs when held aloft by the tail.
Statistical analysis
Data analysis was performed using Prism 4.0 software (GraphPad). Raw data were first analyzed using one-way ANOVA, and then Tukey’s post-test was performed to compare all treatment groups. Differences with *P < 0.05, **P < 0.01, and ***P < 0.001 were considered significantly different.
| Abbreviations: | ||
| RML | = | Rocky Mountain Laboratories |
| NBH | = | normal brain homogenate |
| PKCδ | = | protein kinase Cδ |
| COSC | = | cerebellar organotypic slice cultures |
| TSE | = | transmissible spongiform encephalopathies |
| Thr | = | threonine |
| Tyr | = | tyrosine |
| POSCA | = | prion organotypic slice culture assay |
Acknowledgments
This was supported by the National Institutes of Health grants, ES19267, ES10586 and NS074443. The W. Eugene and Linda Lloyd Endowed Chair is also acknowledged. We also thank Mr. Gary Zenitsky for assistance in preparing this manuscript.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Related Research Data
References
- Aguzzi A, Baumann F, Bremer J. The prion’s elusive reason for being. Annu Rev Neurosci 2008; 31:439 - 77; http://dx.doi.org/10.1146/annurev.neuro.31.060407.125620; PMID: 18558863
- Martin DP, Anantharam V, Jin H, Witte T, Houk R, Kanthasamy A, Kanthasamy AG. Infectious prion protein alters manganese transport and neurotoxicity in a cell culture model of prion disease. Neurotoxicology 2011; 32:554 - 62; http://dx.doi.org/10.1016/j.neuro.2011.07.008; PMID: 21871919
- Prusiner SB. Molecular biology and pathogenesis of prion diseases. Trends Biochem Sci 1996; 21:482 - 7; http://dx.doi.org/10.1016/S0968-0004(96)10063-3; PMID: 9009832
- Anantharam V, Kanthasamy A, Choi CJ, Martin DP, Latchoumycandane C, Richt JA, Kanthasamy AG. Opposing roles of prion protein in oxidative stress- and ER stress-induced apoptotic signaling. Free Radic Biol Med 2008; 45:1530 - 41; http://dx.doi.org/10.1016/j.freeradbiomed.2008.08.028; PMID: 18835352
- Collinge J, Whittington MA, Sidle KC, Smith CJ, Palmer MS, Clarke AR, Jefferys JG. Prion protein is necessary for normal synaptic function. Nature 1994; 370:295 - 7; http://dx.doi.org/10.1038/370295a0; PMID: 8035877
- Kanthasamy AG, Choi C, Jin H, Harischandra DS, Anantharam V, Kanthasamy A. Effect of divalent metals on the neuronal proteasomal system, prion protein ubiquitination and aggregation. Toxicol Lett 2012; 214:288 - 95; http://dx.doi.org/10.1016/j.toxlet.2012.09.008; PMID: 22995398
- Choi CJ, Kanthasamy A, Anantharam V, Kanthasamy AG. Interaction of metals with prion protein: possible role of divalent cations in the pathogenesis of prion diseases. Neurotoxicology 2006; 27:777 - 87; http://dx.doi.org/10.1016/j.neuro.2006.06.004; PMID: 16860868
- Prusiner SB, Groth D, Serban A, Koehler R, Foster D, Torchia M, Burton D, Yang SL, DeArmond SJ. Ablation of the prion protein (PrP) gene in mice prevents scrapie and facilitates production of anti-PrP antibodies. Proc Natl Acad Sci U S A 1993; 90:10608 - 12; http://dx.doi.org/10.1073/pnas.90.22.10608; PMID: 7902565
- Guentchev M, Voigtländer T, Haberler C, Groschup MH, Budka H. Evidence for oxidative stress in experimental prion disease. Neurobiol Dis 2000; 7:270 - 3; http://dx.doi.org/10.1006/nbdi.2000.0290; PMID: 10964599
- Kim JI, Choi SI, Kim NH, Jin JK, Choi EK, Carp RI, Kim YS. Oxidative stress and neurodegeneration in prion diseases. Ann N Y Acad Sci 2001; 928:182 - 6; http://dx.doi.org/10.1111/j.1749-6632.2001.tb05648.x; PMID: 11795509
- Brown DR. Neurodegeneration and oxidative stress: prion disease results from loss of antioxidant defence. Folia Neuropathol 2005; 43:229 - 43; PMID: 16416388
- Kanthasamy A, Jin H, Mehrotra S, Mishra R, Kanthasamy A, Rana A. Novel cell death signaling pathways in neurotoxicity models of dopaminergic degeneration: relevance to oxidative stress and neuroinflammation in Parkinson’s disease. Neurotoxicology 2010; 31:555 - 61; http://dx.doi.org/10.1016/j.neuro.2009.12.003; PMID: 20005250
- Kaul S, Anantharam V, Yang Y, Choi CJ, Kanthasamy A, Kanthasamy AG. Tyrosine phosphorylation regulates the proteolytic activation of protein kinase Cdelta in dopaminergic neuronal cells. J Biol Chem 2005; 280:28721 - 30; http://dx.doi.org/10.1074/jbc.M501092200; PMID: 15961393
- Lin M, Chandramani-Shivalingappa P, Jin H, Ghosh A, Anantharam V, Ali S, Kanthasamy AG, Kanthasamy A. Methamphetamine-induced neurotoxicity linked to ubiquitin-proteasome system dysfunction and autophagy-related changes that can be modulated by protein kinase C delta in dopaminergic neuronal cells. Neuroscience 2012; 210:308 - 32; http://dx.doi.org/10.1016/j.neuroscience.2012.03.004; PMID: 22445524
- Saminathan H, Asaithambi A, Anantharam V, Kanthasamy AG, Kanthasamy A. Environmental neurotoxic pesticide dieldrin activates a non receptor tyrosine kinase to promote PKCδ-mediated dopaminergic apoptosis in a dopaminergic neuronal cell model. Neurotoxicology 2011; 32:567 - 77; http://dx.doi.org/10.1016/j.neuro.2011.06.009; PMID: 21801747
- Mewes A, Franke H, Singer D. Organotypic brain slice cultures of adult transgenic P301S mice--a model for tauopathy studies. PLoS One 2012; 7:e45017; http://dx.doi.org/10.1371/journal.pone.0045017; PMID: 22984603
- Cho S, Wood A, Bowlby MR. Brain slices as models for neurodegenerative disease and screening platforms to identify novel therapeutics. Curr Neuropharmacol 2007; 5:19 - 33; http://dx.doi.org/10.2174/157015907780077105; PMID: 18615151
- Falsig J, Julius C, Margalith I, Schwarz P, Heppner FL, Aguzzi A. A versatile prion replication assay in organotypic brain slices. Nat Neurosci 2008; 11:109 - 17; http://dx.doi.org/10.1038/nn2028; PMID: 18066056
- Falsig J, Sonati T, Herrmann US, Saban D, Li B, Arroyo K, Ballmer B, Liberski PP, Aguzzi A. Prion pathogenesis is faithfully reproduced in cerebellar organotypic slice cultures. PLoS Pathog 2012; 8:e1002985; http://dx.doi.org/10.1371/journal.ppat.1002985; PMID: 23133383
- Bolton DC, McKinley MP, Prusiner SB. Identification of a protein that purifies with the scrapie prion. Science 1982; 218:1309 - 11; http://dx.doi.org/10.1126/science.6815801; PMID: 6815801
- Polymenidou M, Moos R, Scott M, Sigurdson C, Shi YZ, Yajima B, Hafner-Bratkovic I, Jerala R, Hornemann S, Wuthrich K, et al. The POM monoclonals: a comprehensive set of antibodies to non-overlapping prion protein epitopes. PLoS One 2008; 3:e3872; http://dx.doi.org/10.1371/journal.pone.0003872; PMID: 19060956
- Sigurdson CJ, Joshi-Barr S, Bett C, Winson O, Manco G, Schwarz P, Rülicke T, Nilsson KP, Margalith I, Raeber A, et al. Spongiform encephalopathy in transgenic mice expressing a point mutation in the β2-α2 loop of the prion protein. J Neurosci 2011; 31:13840 - 7; http://dx.doi.org/10.1523/JNEUROSCI.3504-11.2011; PMID: 21957246
- Jin H, Kanthasamy A, Anantharam V, Rana A, Kanthasamy AG. Transcriptional regulation of pro-apoptotic protein kinase Cdelta: implications for oxidative stress-induced neuronal cell death. J Biol Chem 2011; 286:19840 - 59; http://dx.doi.org/10.1074/jbc.M110.203687; PMID: 21467032
- Gordon R, Anantharam V, Kanthasamy AG, Kanthasamy A. Proteolytic activation of proapoptotic kinase protein kinase Cδ by tumor necrosis factor α death receptor signaling in dopaminergic neurons during neuroinflammation. J Neuroinflammation 2012; 9:82; http://dx.doi.org/10.1186/1742-2094-9-82; PMID: 22540228
- Kanthasamy AG, Kitazawa M, Kaul S, Yang Y, Lahiri DK, Anantharam V, Kanthasamy A. Proteolytic activation of proapoptotic kinase PKCdelta is regulated by overexpression of Bcl-2: implications for oxidative stress and environmental factors in Parkinson’s disease. Ann N Y Acad Sci 2003; 1010:683 - 6; http://dx.doi.org/10.1196/annals.1299.125; PMID: 15033812
- Pamplona R, Naudí A, Gavín R, Pastrana MA, Sajnani G, Ilieva EV, Del Río JA, Portero-Otín M, Ferrer I, Requena JR. Increased oxidation, glycoxidation, and lipoxidation of brain proteins in prion disease. Free Radic Biol Med 2008; 45:1159 - 66; http://dx.doi.org/10.1016/j.freeradbiomed.2008.07.009; PMID: 18703134
- Freixes M, Rodríguez A, Dalfó E, Ferrer I. Oxidation, glycoxidation, lipoxidation, nitration, and responses to oxidative stress in the cerebral cortex in Creutzfeldt-Jakob disease. Neurobiol Aging 2006; 27:1807 - 15; http://dx.doi.org/10.1016/j.neurobiolaging.2005.10.006; PMID: 16310893
- Yuan F, Yang L, Zhang Z, Wu W, Zhou X, Yin X, Zhao D. Cellular prion protein (PrPC) of the neuron cell transformed to a PK-resistant protein under oxidative stress, comprising main mitochondrial damage in prion diseases. J Mol Neurosci 2013; 51:219 - 24; http://dx.doi.org/10.1007/s12031-013-0008-6; PMID: 23715697
- Jin H, Kanthasamy A, Ghosh A, Anantharam V, Kalyanaraman B, Kanthasamy AG. Mitochondria-targeted antioxidants for treatment of Parkinson’s disease: Preclinical and clinical outcomes. Biochim Biophys Acta 2013; http://dx.doi.org/10.1016/j.bbadis.2013.09.007; PMID: 24060637
- Latchoumycandane C, Anantharam V, Jin H, Kanthasamy A, Kanthasamy A. Dopaminergic neurotoxicant 6-OHDA induces oxidative damage through proteolytic activation of PKCδ in cell culture and animal models of Parkinson’s disease. Toxicol Appl Pharmacol 2011; 256:314 - 23; http://dx.doi.org/10.1016/j.taap.2011.07.021; PMID: 21846476
- Li W, Mischak H, Yu JC, Wang LM, Mushinski JF, Heidaran MA, Pierce JH. Tyrosine phosphorylation of protein kinase C-delta in response to its activation. J Biol Chem 1994; 269:2349 - 52; PMID: 7507923
- Le Good JA, Ziegler WH, Parekh DB, Alessi DR, Cohen P, Parker PJ. Protein kinase C isotypes controlled by phosphoinositide 3-kinase through the protein kinase PDK1. Science 1998; 281:2042 - 5; http://dx.doi.org/10.1126/science.281.5385.2042; PMID: 9748166
- Yang Q, Hashizume Y, Yoshida M, Wang Y. Neuropathological study of cerebellar degeneration in prion disease. Neuropathology 1999; 19:33 - 9; http://dx.doi.org/10.1046/j.1440-1789.1999.00212.x; PMID: 19519645
- Carimalo J, Cronier S, Petit G, Peyrin JM, Boukhtouche F, Arbez N, Lemaigre-Dubreuil Y, Brugg B, Miquel MC. Activation of the JNK-c-Jun pathway during the early phase of neuronal apoptosis induced by PrP106-126 and prion infection. Eur J Neurosci 2005; 21:2311 - 9; http://dx.doi.org/10.1111/j.1460-9568.2005.04080.x; PMID: 15932590
- Bourteele S, Oesterle K, Weinzierl AO, Paxian S, Riemann M, Schmid RM, Planz O. Alteration of NF-kappaB activity leads to mitochondrial apoptosis after infection with pathological prion protein. Cell Microbiol 2007; 9:2202 - 17; http://dx.doi.org/10.1111/j.1462-5822.2007.00950.x; PMID: 17573907
- Aguzzi A, Heikenwalder M. Pathogenesis of prion diseases: current status and future outlook. Nat Rev Microbiol 2006; 4:765 - 75; http://dx.doi.org/10.1038/nrmicro1492; PMID: 16980938
- Bessen RA, Robinson CJ, Seelig DM, Watschke CP, Lowe D, Shearin H, Martinka S, Babcock AM. Transmission of chronic wasting disease identifies a prion strain causing cachexia and heart infection in hamsters. PLoS One 2011; 6:e28026; http://dx.doi.org/10.1371/journal.pone.0028026; PMID: 22174765
- Castilla J, Morales R, Saá P, Barria M, Gambetti P, Soto C. Cell-free propagation of prion strains. EMBO J 2008; 27:2557 - 66; http://dx.doi.org/10.1038/emboj.2008.181; PMID: 18800058
- Collins S, McLean CA, Masters CL. Gerstmann-Sträussler-Scheinker syndrome,fatal familial insomnia, and kuru: a review of these less common human transmissible spongiform encephalopathies. J Clin Neurosci 2001; 8:387 - 97; http://dx.doi.org/10.1054/jocn.2001.0919; PMID: 11535002
- O’Shea M, Maytham EG, Linehan JM, Brandner S, Collinge J, Lloyd SE. Investigation of mcp1 as a quantitative trait gene for prion disease incubation time in mouse. Genetics 2008; 180:559 - 66; http://dx.doi.org/10.1534/genetics.108.090894; PMID: 18716327
- Zhang D, Anantharam V, Kanthasamy A, Kanthasamy AG. Neuroprotective effect of protein kinase C delta inhibitor rottlerin in cell culture and animal models of Parkinson’s disease. J Pharmacol Exp Ther 2007; 322:913 - 22; http://dx.doi.org/10.1124/jpet.107.124669; PMID: 17565007
- Falsig J, Aguzzi A. The prion organotypic slice culture assay--POSCA. Nat Protoc 2008; 3:555 - 62; http://dx.doi.org/10.1038/nprot.2008.13; PMID: 18388937
- Kanthasamy AG, Kitazawa M, Kanthasamy A, Anantharam V. Role of proteolytic activation of protein kinase Cdelta in oxidative stress-induced apoptosis. Antioxid Redox Signal 2003; 5:609 - 20; http://dx.doi.org/10.1089/152308603770310275; PMID: 14580317
- Jin H, Kanthasamy A, Ghosh A, Yang Y, Anantharam V, Kanthasamy AG. α-Synuclein negatively regulates protein kinase Cδ expression to suppress apoptosis in dopaminergic neurons by reducing p300 histone acetyltransferase activity. J Neurosci 2011; 31:2035 - 51; http://dx.doi.org/10.1523/JNEUROSCI.5634-10.2011; PMID: 21307242
- Sinclair L, Lewis V, Collins SJ, Haigh CL. Cytosolic caspases mediate mislocalised SOD2 depletion in an in vitro model of chronic prion infection. Dis Model Mech 2013; 6:952 - 63; http://dx.doi.org/10.1242/dmm.010678; PMID: 23580200
- Sonati T, Reimann RR, Falsig J, Baral PK, O’Connor T, Hornemann S, Yaganoglu S, Li B, Herrmann US, Wieland B, et al. The toxicity of antiprion antibodies is mediated by the flexible tail of the prion protein. Nature 2013; 501:102 - 6; http://dx.doi.org/10.1038/nature12402; PMID: 23903654
- Kikkawa U, Matsuzaki H, Yamamoto T. Protein kinase C delta (PKC delta): activation mechanisms and functions. J Biochem 2002; 132:831 - 9; http://dx.doi.org/10.1093/oxfordjournals.jbchem.a003294; PMID: 12473183
- Anantharam V, Kitazawa M, Wagner J, Kaul S, Kanthasamy AG. Caspase-3-dependent proteolytic cleavage of protein kinase Cdelta is essential for oxidative stress-mediated dopaminergic cell death after exposure to methylcyclopentadienyl manganese tricarbonyl. J Neurosci 2002; 22:1738 - 51; PMID: 11880503
- Kitazawa M, Anantharam V, Kanthasamy AG. Dieldrin induces apoptosis by promoting caspase-3-dependent proteolytic cleavage of protein kinase Cdelta in dopaminergic cells: relevance to oxidative stress and dopaminergic degeneration. Neuroscience 2003; 119:945 - 64; http://dx.doi.org/10.1016/S0306-4522(03)00226-4; PMID: 12831855
- Reyland ME. Protein kinase Cdelta and apoptosis. Biochem Soc Trans 2007; 35:1001 - 4; http://dx.doi.org/10.1042/BST0351001; PMID: 17956263
- Yun SW, Gerlach M, Riederer P, Klein MA. Oxidative stress in the brain at early preclinical stages of mouse scrapie. Exp Neurol 2006; 201:90 - 8; http://dx.doi.org/10.1016/j.expneurol.2006.03.025; PMID: 16806186
- Merchenthaler I, Liposits Z, Reid JJ, Wetsel WC. Light and electron microscopic immunocytochemical localization of PKC delta immunoreactivity in the rat central nervous system. J Comp Neurol 1993; 336:378 - 99; http://dx.doi.org/10.1002/cne.903360306; PMID: 8263228
- Barmack NH, Qian Z, Yoshimura J. Regional and cellular distribution of protein kinase C in rat cerebellar Purkinje cells. J Comp Neurol 2000; 427:235 - 54; http://dx.doi.org/10.1002/1096-9861(20001113)427:2<235::AID-CNE6>3.0.CO;2-6; PMID: 11054691
- Cooper SA, Murray KL, Heath CA, Will RG, Knight RS. Sporadic Creutzfeldt-Jakob disease with cerebellar ataxia at onset in the UK. J Neurol Neurosurg Psychiatry 2006; 77:1273 - 5; http://dx.doi.org/10.1136/jnnp.2006.088930; PMID: 16835290
- Glatzel M, Stoeck K, Seeger H, Lührs T, Aguzzi A. Human prion diseases: molecular and clinical aspects. Arch Neurol 2005; 62:545 - 52; http://dx.doi.org/10.1001/archneur.62.4.545; PMID: 15824251
- Deacon RM. Measuring the strength of mice. J Vis Exp 2013; PMID: 23770643
- Chou AH, Yeh TH, Ouyang P, Chen YL, Chen SY, Wang HL. Polyglutamine-expanded ataxin-3 causes cerebellar dysfunction of SCA3 transgenic mice by inducing transcriptional dysregulation. Neurobiol Dis 2008; 31:89 - 101; http://dx.doi.org/10.1016/j.nbd.2008.03.011; PMID: 18502140
- Guyenet SJ, Furrer SA, Damian VM, Baughan TD, La Spada AR, Garden GA. A simple composite phenotype scoring system for evaluating mouse models of cerebellar ataxia. J Vis Exp 2010; http://dx.doi.org/10.3791/1787; PMID: 20495529
- Ghosh A, Saminathan H, Kanthasamy A, Anantharam V, Jin H, Sondarva G, Harischandra DS, Qian Z, Rana A, Kanthasamy AG. The peptidyl-prolyl isomerase Pin1 up-regulation and proapoptotic function in dopaminergic neurons: relevance to the pathogenesis of Parkinson disease. J Biol Chem 2013; 288:21955 - 71; http://dx.doi.org/10.1074/jbc.M112.444224; PMID: 23754278
- Kanthasamy AG, Anantharam V, Zhang D, Latchoumycandane C, Jin H, Kaul S, Kanthasamy A. A novel peptide inhibitor targeted to caspase-3 cleavage site of a proapoptotic kinase protein kinase C delta (PKCdelta) protects against dopaminergic neuronal degeneration in Parkinson’s disease models. Free Radic Biol Med 2006; 41:1578 - 89; http://dx.doi.org/10.1016/j.freeradbiomed.2006.08.016; PMID: 17045926
- Miyamoto A, Nakayama K, Imaki H, Hirose S, Jiang Y, Abe M, Tsukiyama T, Nagahama H, Ohno S, Hatakeyama S, et al. Increased proliferation of B cells and auto-immunity in mice lacking protein kinase Cdelta. Nature 2002; 416:865 - 9; http://dx.doi.org/10.1038/416865a; PMID: 11976687
- Mallucci GR, White MD, Farmer M, Dickinson A, Khatun H, Powell AD, Brandner S, Jefferys JG, Collinge J. Targeting cellular prion protein reverses early cognitive deficits and neurophysiological dysfunction in prion-infected mice. Neuron 2007; 53:325 - 35; http://dx.doi.org/10.1016/j.neuron.2007.01.005; PMID: 17270731
- Guenther K, Deacon RM, Perry VH, Rawlins JN. Early behavioural changes in scrapie-affected mice and the influence of dapsone. Eur J Neurosci 2001; 14:401 - 9; http://dx.doi.org/10.1046/j.0953-816x.2001.01645.x; PMID: 11553290