
Abstract
It was shown previously that truncated molecules of prion protein can be found in brains of patients with some types of transmissible spongiform encephalopathy. One such molecule, PrP226*, is a fragment of prion protein, truncated at Tyr226. It was found to be present in aggregates, from which it can be released using chaotropic salts. In this study we investigated the distribution of PrP226* in Creutzfeldt–Jakob disease affected human brain, employing the mAb V5B2, specifically recognizing this fragment. The results show that PrP226* is not evenly distributed among different regions of human brain. Among brain regions analyzed, the fragment was found most likely to be accumulated in the cerebellum. Its distribution correlates with the distribution of PrPSc.
Introduction
Prion protein (PrP) is a glycoprotein, bound to the cell membrane through a glycophosphatidylinositol (GPI) anchor, and is expressed in all mammalian cells with the highest levels of expression in neurons.Citation1 Cellular form of PrP (PrPC) can undergo posttranslational modifications that shift secondary structure of the molecule from mostly α-helical to β-sheeted, changing the properties of the protein drastically.Citation2 Altered molecules, named PrPSc, are prone to aggregation and are partially resistant to digestion with proteinase K. The enzyme cleaves off a part of molecule from the N-terminus, leaving the core fragment of 19 or 21 kDa (PrPres), whereas PrPC molecules are totally degraded.Citation3 The size of the PrPres along with the genotype on polymorph codon 129 is the basis for the molecular classification of sCJD.Citation4
Prion diseases, also named transmissible spongiform encephalopathies (TSEs), are rare fatal neurodegenerative diseases that affect humans and animals. Sporadic Creutzfeldt-Jakob disease (sCJD) is the most common form of human TSE, other forms are much less frequent. A new variant of CJD (vCJD), that first appeared in Great Britain in 1995,Citation4 was connected to consumption of meat products originating from cattle infected with bovine spongiform encephalopathy.
Besides PrPSc, the presence of truncated PrP molecules in brain has been described in sCJD as well as in genetic form of TSE named Gerstmann–Sträussler–Scheinker syndrome.Citation5-Citation7 Our group reported on the discovery of one such fragment, as well as the production and characterization of a mAb V5B2 specifically recognizing it.Citation8,Citation9 mAb V5B2 was prepared by immunization of BALB/c mice with a peptide from the sequence of human PrP (amino acid residues 214–226).Citation8 The mAb was especially interesting, because it discriminated between CJD and non-CJD human brain tissue in dot-blot, immunohistochemistry and western blot.Citation8,Citation10 Surprisingly, it turned out that the V5B2 does not discriminate between conformational isoforms of PrPC and PrPSc, but binds exclusively the truncated form of PrP ending with Tyr226.Citation9 It was shown that it binds with high affinity to recHuPrP(23–226), while no reaction is observed with recHuPrP(23–231).Citation9 The epitope was determined by alanine scanning and phage display. Substitution of C-terminal Tyr226 as well as the addition of one amino acid C-terminally of Tyr226 completely abolished the binding of V5B2, indicating crucial role of this amino acid residue for binding.Citation9 The fragment is C-terminally truncated and thus an anchorless PrP molecule that ends with the residue Tyr226 and was therefore named PrP226*.Citation9 A sandwich immunoassay for detection of PrP226* in brain homogenates using V5B2 was developed,Citation10 employing denaturation to unpack the PrP226* molecules from the aggregates. This kind of approach was first introduced by Serban et al.,Citation11 and since then often used for unpacking PrPSc molecules from aggregates. Upon the denaturation with chaotropic salts, PrPSc aggregates are loosened and epitopes, previously unavailable for interaction with certain Ab, are revealed. The ratio between signal of denatured and non-denatured sample is the measure for the epitopes revealed and indicates the amount of detected molecules in the sample.Citation12 For the purpose of detecting PrP226*, mAb V5B2 was used instead of an antibody recognizing the whole PrP molecule.
In this study we assessed the presence of PrP226* in five different human brain regions of several sCJD cases and controls. We present for the first time a systematic analysis of distribution of this fragment in sCJD-affected human brain.
Results and Discussion
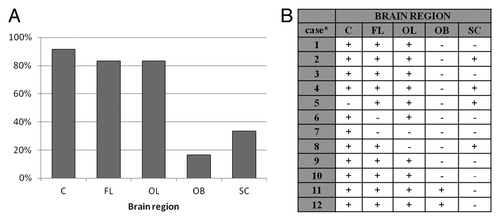
The amount of PrP226*, represented as D/N ratio, varied considerably between different brain regions of individual cases as well as between the same brain regions of different cases (). There was an obvious difference in amount of PrP226* between cerebellum (C), frontal lobe (FL) and occipital lobe (OL) samples on one hand and olfactory bulb (OB) and spinal cord (SC) samples on the other, the former containing much higher amounts of the fragment. In 5/12 cases the amount of PrP226* was the highest in C, in 4/12 cases in FL and in 3/12 cases in OL. OB and SC were never the regions with the highest amount of PrP226*. We could define as positive 11/12 C samples (92%), 10/12 OL samples (83%), 10/12 FL (83%) whereas only 4/12 SC samples (33%) and 2/12 OB sample (17%) (). For all 12 sCJD cases at least one of five tested regions was positive and at least one was negative (). For all control samples D/N ratio was below 1 ().
Figure 1. Average D/N ratios for all samples. (A) D/N ratios for sCJD samples. C (black); FL (dark-gray); OL (gray); OB (light-gray); SC (white). (B) D/N ratios for non-CJD samples. Dark-gray columns represent NC (13–19) and light-gray columns represent NNC (20–30). The data was obtained from PrP226* assay. Sequential numbers of samples are as in . C, cerebellum; FL, frontal lobe; OL, occipital lobe; OB, olfactory bulb; SC, spinal cord.
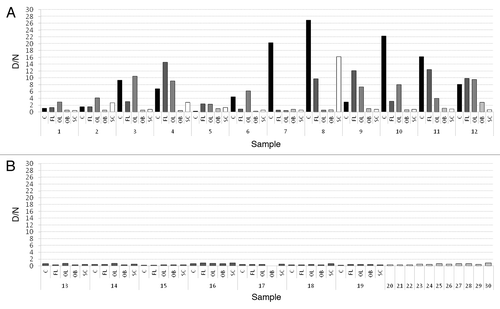
Figure 2. (A) Percentage of samples from certain brain region of 12 sCJD cases, which were defined as positive. (B) Results of PrP226* assay for all sCJD cases. Sequential numbers of samples are as in
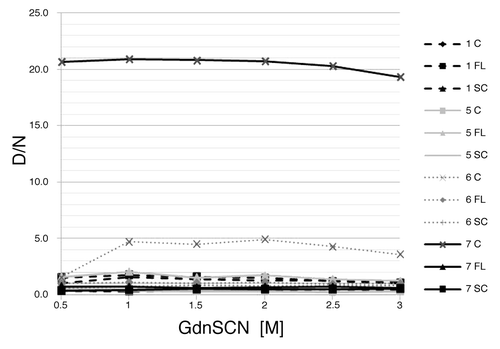
We obtained denaturation profiles of samples with high or low D/N ratios to make sure that the ratios correlate with the amount of PrP226* and not with the susceptibility to denaturation (). For none of the tested samples did D/N ratio increase with the increasing denaturant concentration, indicating that low D/N ratios are not a consequence of resistance to denaturation.
Figure 3. Denaturation profiles. D/N ratios are ploted as the function of the GdnSCN concentration. Sequential numbers of samples are as in . C, cerebellum; FL, frontal lobe; SC, spinal cord.
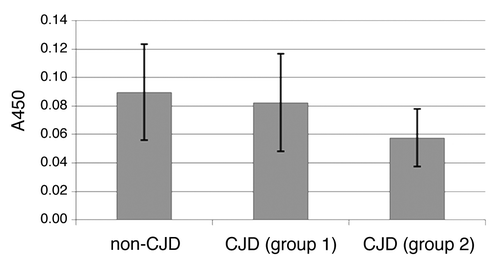
Measurements of A450 of non-denatured samples showed that not all of PrP226* in samples from sCJD cases is entangled in aggregates. Furthermore, free PrP226* was found to be present in minute amounts also in non-CJD brains. Closer examination of the amounts of free PrP226* revealed that samples, defined as positive, contained lower amounts of free PrP226* than sCJD samples, defined as negative, and control samples (). The difference in the amounts of free PrP226* between CJD samples, defined as positive, and CJD samples, defined as negative, as well as the difference in the amounts of free PrP226* between CJD samples, defined as positive, and negative controls, was statistically significant (P < 0.002 and P < 0.0001 respectfully), determined with Kruskal-Wallis test. These data support the hypothesis that free PrP226* is incorporated into the aggregates as they are formed.
Figure 4. Comparison of A450 values of non-denatured samples (all brain regions included). The first bar represents the average value of non-denatured non-CJD samples (NC, NNC). sCJD samples were divided in two groups – samples, that were defined as negative (D/n < 1.2) were included in group 1, samples, that were defined as positive (D/n > 1) were included in group 2. The second bar represents the average value of non-denatured sCJD samples from group 1 and the third bar represents the average value of non-denatured sCJD samples from group 2.
The comparison of the results of PrP226* and PrPSc assay showed that the amount of PrP226* correlates with the amount of PrPSc in the sample as detected in the assay, where mAb 3F4 was used as capturing Ab in 3F4-E12/2 sandwich. Since we systematically analyzed multiple brain regions of each individual case, we additionally found out, that the distribution of PrP226* follows the pattern of PrPSc distribution (data not shown). This observation was not surprising in the view of our hypothesis that PrP226* is a part of PrPSc aggregates and released from them along with PrPSc upon denaturation.
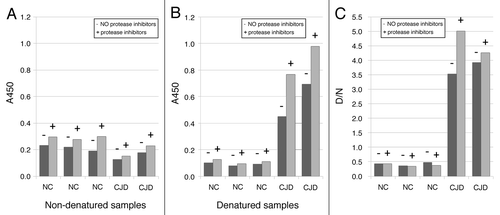
PrP226* is a fragment of PrP, ending with the residue 226. It could therefore be generated from the whole PrP molecule by proteolitic degradation, especially since no protease inhibitors were added into the homogenization buffer. To test the possibility that PrP226* was generated ex-vivo during the homogenization process, multiple CJD and NC samples were homogenized simultaneously in the presence and absence of protease inhibitors. Measurements of A450 () revealed slight increase in the amounts of PrP226* when protease inhibitors were used, indicating that PrP226* is degraded rather than generated during homogenization without protease inhibitors. The loss of PrP226* due to the proteolitic activity did not influence D/N ratios crucially. Due to the limited amounts of samples available, we decided not to use protease inhibitors in homogenization buffer, what enabled the usage of the same homogenates also for other experiments, such as proteinase K digestion. This data suggest that PrP226* is a naturally occurring fragment of PrP, generated in healthy brain in minute amount and accumulating during the disease. The fragment could also be further degraded after homogenization. In order to determine whether the amount of PrP226* in the sample changes after multiple defrosting, aliquots of sCJD and non-CJD brain homogenates were subjected to multiple rounds of thawing and freezing cycles. Only minimal changes in measured A450 after serial thawing of samples were found, suggesting that the amounts of PrP226* epitopes remained constant during the treatment (data not shown). The conclusion was made, that the PrP226* assay, employing mAb V5B2 and E12/2 is a very robust one. This finding cannot be generalized as different epitopes may undergo different changes.
Figure 5. Influence of protease inhibitors in homogenization buffer on the amount of PrP226*. Samples were homogenized in buffer with (+) or without (-) of protease inhibitors. A450 values are shown for non-denatured (A) and denatured (B) samples.
In this study, we analyzed the distribution of PrP226* in five different regions of human brain. This is the first study addressing the distribution of this fragment in human brain. The fragment was found not to be evenly distributed among different regions of sCJD-affected human brain and no pattern of distribution was found. Cerebellum was identified as the region where PrP226* is most likely to be accumulated, followed by occipital lobe and frontal lobe ().
Whereas nothing has been known so far about the distribution of PrP226* in human brain, the regional heterogeneity of PrPSc as well as PrPC in human and animal brain has been addressed in several studies.Citation13-Citation18 Choi et al.Citation13 employed conformation dependent immunoassay to investigate the abundance of PrPSc in two different brain regions, namely frontal cortex and cerebellum in cases of MM at codon 129 sCJD. They showed that PrPSc was most abundant in frontal cortex and less in cerebellum. These findings are in agreement with the former study of Schoch et al.,Citation17 who analyzed the distribution of PrPres in nine different brain regions of MM, VV and MV sCJD cases, including frontal cortex, occipital cortex and cerebellum. In MM cases, the most PrPres was found in cortical regions and less in cerebellum, whereas in VV cases the situation was the opposite, namely cerebellum was the region with the highest amounts of PrPres. It should be taken into consideration that in all mentioned studies, Abs used were directed against epitopes N-terminally of Tyr226. That means that detected molecules could among others also be PrP226*.
Among our studied cases, we had a single VV case in which substantial amounts of PrPSc as well as PrP226* were found only in cerebellum, what is in agreement with before mentioned studies.Citation13,Citation17 VV type case was also the only analyzed case with such an extreme difference in the amount of PrP226* between brain regions, indicating that genotype may influence not only PrPSc, but also PrP226* distribution. For 10 MM cases the results were not uniform, nevertheless, in the majority of cases higher amounts of PrPSc and PrP226* were found in cortical regions compared with cerebellum, what is in accordance with the findings of Choi et al.Citation13
Since PrP226* is a PrP molecule, truncated at the very C-terminus, it lacks the GPI anchor. Until recently it was believed that GPI anchor is an essential feature for conversion of PrPC to PrPSc, but this assumption was rebutted when it was shown that transgenic mice, expressing only anchorless PrP, can be efficiently infected with scrapieCitation19,Citation20 and that even spontaneous disease can occur in time.Citation21 Moreover, Jansen et al.Citation22 described two unusually presenting GSS cases in which nonsense mutations at one locus at the C-terminus of PrP resulted in the expression of anchorless PrP. In one case the protein ended with Tyr226 what means that abundant amounts of PrP226* were expressed and accumulated in patients brain. With one allele expressing full-length, GPI-anchored PrP, the expression of PrP226* seems to be the cause of the disease. The regional distribution of PrP226* was not addressed and the distribution of PrPSc deposits was assessed according to IHC so the comparison with our results is not possible.
It is not clear at this point how many different PrP fragments, among which PrP226* is quite abundant, can be generated in human brain. They are an important feature in presentation of TSEs but their exact role has not been determined yet. From the information gathered so far on PrP226* it is clear that accumulation of this fragment is characteristic for at least some types of human TSEs (sCJD, vCJD, GSS)Citation8,Citation22 and also for BSE and scrapie (unpublished data). Since its distribution in sCJD follows that of PrPSc aggregates, it could be considered as surrogate marker for PrPSc. As an anchorless PrP molecule, PrP226* is likely to be present in body fluids such as cerebrospinal fluid, urine and blood to which the development of future diagnostic of human TSE is focused.
Materials and Methods
Consent for using the samples was obtained from National Medical Ethics Committee. Human brain tissue samples of 12 sCJD cases (5 males and 7 females) and 7 clinically suspected sCJD patients, who were disproved after post mortem examination by applying immunohistochemistry on PrPSc, called neurological controls (NC) (3 males and 4 females) were obtained from the Institute of Pathology, Medical Faculty, University of Ljubljana. For each sCJD case and NC, samples from five brain regions (cerebellum (C), frontal lobe (FL), occipital lobe (OL), olfactory bulb (OB) and spinal cord (SC)) were analyzed, except for one NC for which olfactory bulb tissue was not available. In ten cases of sCJD the genotype of the codon 129 was MM, in one case VV and in one case MV. Additionally, 11 cerebellar samples of patients (4 males and 7 females) with no neurodegenerative disease at time of death were analyzed (non-neurological controls, NNC). Detailed information on patients can be found in .
Table 1. Summary of data on patients included in the study
10% (w/v) brain tissue homogenates were prepared in homogenization buffer (10 mM Tris buffered saline, pH 7.5, containing 100 mM NaCl, 10 mM EDTA, 0.5% NP-40 and 0.5% sodium deoxycholate) using zirconium oxide 0.5 mm beads (Next Advance) and Tissue Lyser LT (Qiagen), aliquoted and frozen at -80 °C until use. For the test, each sample was prepared under denaturing and non-denaturing conditions. For denaturation, aliquots of 10% brain homogenates were mixed with equal volume of 3 M GdnSCN and incubated for 15 min at 60 °C with shaking. Nondenatured samples were mixed with equal volume of 50 mM TRIS-HCl instead of denaturant and left at room temperature for 15 min. Before loading to the microtiter plate, sample mixtures were diluted 10 times. For obtaining the denaturation profiles, chosen samples were denatured with increasing concentrations of GdnSCN, ranging from 0.5 M to 3 M GdnSCN in sample mixture. Final concentration of GdnSCN on microtiter plate never exceeded 0.3 M to avoid the negative influence of denaturant on coated Ab.
The immunoassay, used in this study, was based on the assay, developed for the detection of PrP226*,Citation10 however some important changes were introduced. The DELFIA protocol was substituted with standard ELISA. Unlike in previously described PrP226* assay, here mAb E12/223 was used as detecting Ab. Its epitope was located at C-terminal end of helix 1 of human PrP with His155 being crucial for binding.Citation23 Microtiter plate was coated with mAb V5B2 (5 μg ml−1). Prepared samples were loaded and incubated for 90 min. Plate was then incubated with biotinylated mAb E12/2 and subsequently with HRP-labeled avidin. To develop color reaction, TMB was added and microtiter plate was incubated for 10 min. Reaction was terminated with the addition of 0.1 M H2SO4. The absorbance at 450 nm (A450) of denatured samples (D) and of non-denatured samples (N) was measured. The ratios between D and N signals were calculated (D/N ratios). To measure PrPSc, the microtiter plate was coated with mAb 3F4 (Covance) at concentration of 0.8 μg ml−1. All the other procedures were as in the PrP226* assay.
PrP226* assay was performed at least two times for all samples, except for 5 olfactory bulb samples (2, 5, 13, 14, and 17) and one spinal cord sample (18) that were no longer available after the first measurement. Since the denaturation was used for releasing PrP226* from aggregates, the value of D/N ratio was expected to be high for sCJD samples and 1 or below 1 for non-CJD samples. We defined a sample as positive, if the average D/N ratio was above 1.2 and negative, if the average D/N ratio was below 1.2. The results presented are the average D/N ratios of all the measurements for each sample ().
Disclosure of Potential Conflicts of Interest
The authors declare that there are no conflicts of interest or competing financial interests.
Acknowledgments
The study was financially supported by The Slovenian Research Agency with research program P4–0176 and Ph.D. grant to A.L.
Related Research Data
References
- Oesch B, Westaway D, Wälchli M, McKinley MP, Kent SB, Aebersold R, Barry RA, Tempst P, Teplow DB, Hood LE, et al. A cellular gene encodes scrapie PrP 27-30 protein. Cell 1985; 40:735 - 46; http://dx.doi.org/10.1016/0092-8674(85)90333-2; PMID: 2859120
- Pan KM, Baldwin M, Nguyen J, Gasset M, Serban A, Groth D, Mehlhorn I, Huang Z, Fletterick RJ, Cohen FE, et al. Conversion of alpha-helices into beta-sheets features in the formation of the scrapie prion proteins. Proc Natl Acad Sci U S A 1993; 90:10962 - 6; http://dx.doi.org/10.1073/pnas.90.23.10962; PMID: 7902575
- Meyer RK, McKinley MP, Bowman KA, Braunfeld MB, Barry RA, Prusiner SB. Separation and properties of cellular and scrapie prion proteins. Proc Natl Acad Sci U S A 1986; 83:2310 - 4; http://dx.doi.org/10.1073/pnas.83.8.2310; PMID: 3085093
- Gambetti P, Kong Q, Zou W, Parchi P, Chen SG. Sporadic and familial CJD: classification and characterisation. Br Med Bull 2003; 66:213 - 39; http://dx.doi.org/10.1093/bmb/66.1.213; PMID: 14522861
- Notari S, Strammiello R, Capellari S, Giese A, Cescatti M, Grassi J, Ghetti B, Langeveld JPM, Zou W-Q, Gambetti P, et al. Characterization of truncated forms of abnormal prion protein in Creutzfeldt-Jakob disease. J Biol Chem 2008; 283:30557 - 65; http://dx.doi.org/10.1074/jbc.M801877200; PMID: 18753138
- Zanusso G, Farinazzo A, Prelli F, Fiorini M, Gelati M, Ferrari S, Righetti PG, Rizzuto N, Frangione B, Monaco S. Identification of distinct N-terminal truncated forms of prion protein in different Creutzfeldt-Jakob disease subtypes. J Biol Chem 2004; 279:38936 - 42; http://dx.doi.org/10.1074/jbc.M405468200; PMID: 15247220
- Zou W-Q, Capellari S, Parchi P, Sy M-S, Gambetti P, Chen SG. Identification of novel proteinase K-resistant C-terminal fragments of PrP in Creutzfeldt-Jakob disease. J Biol Chem 2003; 278:40429 - 36; http://dx.doi.org/10.1074/jbc.M308550200; PMID: 12917418
- Curin Serbec V, Bresjanac M, Popovic M, Pretnar Hartman K, Galvani V, Rupreht R, Cernilec M, Vranac T, Hafner I, Jerala R. Monoclonal antibody against a peptide of human prion protein discriminates between Creutzfeldt-Jacob’s disease-affected and normal brain tissue. J Biol Chem 2004; 279:3694 - 8; http://dx.doi.org/10.1074/jbc.M310868200; PMID: 14593100
- Kosmač M, Koren S, Giachin G, Stoilova T, Gennaro R, Legname G, Serbec VČ. Epitope mapping of a PrP(Sc)-specific monoclonal antibody: identification of a novel C-terminally truncated prion fragment. Mol Immunol 2011; 48:746 - 50; http://dx.doi.org/10.1016/j.molimm.2010.11.012; PMID: 21176851
- Dvorakova E, Vranac T, Janouskova O, Černilec M, Koren S, Lukan A, Nováková J, Matej R, Holada K, Čurin Šerbec V. Detection of the GPI-anchorless prion protein fragment PrP226* in human brain. BMC Neurol 2013; 13:126; http://dx.doi.org/10.1186/1471-2377-13-126; PMID: 24063733
- Serban D, Taraboulos A, DeArmond SJ, Prusiner SB. Rapid detection of Creutzfeldt-Jakob disease and scrapie prion proteins. Neurology 1990; 40:110 - 7; http://dx.doi.org/10.1212/WNL.40.1.110; PMID: 1967489
- Safar J, Wille H, Itri V, Groth D, Serban H, Torchia M, Cohen FE, Prusiner SB. Eight prion strains have PrP(Sc) molecules with different conformations. Nat Med 1998; 4:1157 - 65; http://dx.doi.org/10.1038/2654; PMID: 9771749
- Choi YP, Gröner A, Ironside JW, Head MW. Comparison of the level, distribution and form of disease-associated prion protein in variant and sporadic Creutzfeldt-Jakob diseased brain using conformation-dependent immunoassay and Western blot. J Gen Virol 2011; 92:727 - 32; http://dx.doi.org/10.1099/vir.0.026948-0; PMID: 21123539
- Kuczius T, Koch R, Keyvani K, Karch H, Grassi J, Groschup MH. Regional and phenotype heterogeneity of cellular prion proteins in the human brain. Eur J Neurosci 2007; 25:2649 - 55; http://dx.doi.org/10.1111/j.1460-9568.2007.05518.x; PMID: 17466020
- Moya KL, Salès N, Hässig R, Créminon C, Grassi J, Di Giamberardino L. Immunolocalization of the cellular prion protein in normal brain. Microsc Res Tech 2000; 50:58 - 65; http://dx.doi.org/10.1002/1097-0029(20000701)50:1<58::AID-JEMT9>3.0.CO;2-5; PMID: 10871549
- Parchi P, Castellani R, Capellari S, Ghetti B, Young K, Chen SG, Farlow M, Dickson DW, Sima AA, Trojanowski JQ, et al. Molecular basis of phenotypic variability in sporadic Creutzfeldt-Jakob disease. Ann Neurol 1996; 39:767 - 78; http://dx.doi.org/10.1002/ana.410390613; PMID: 8651649
- Schoch G, Seeger H, Bogousslavsky J, Tolnay M, Janzer RC, Aguzzi A, Glatzel M. Analysis of prion strains by PrPSc profiling in sporadic Creutzfeldt-Jakob disease. PLoS Med 2006; 3:e14; http://dx.doi.org/10.1371/journal.pmed.0030014; PMID: 16354106
- Taraboulos A, Jendroska K, Serban D, Yang SL, DeArmond SJ, Prusiner SB. Regional mapping of prion proteins in brain. Proc Natl Acad Sci U S A 1992; 89:7620 - 4; http://dx.doi.org/10.1073/pnas.89.16.7620; PMID: 1354357
- Chesebro B, Trifilo M, Race R, Meade-White K, Teng C, LaCasse R, Raymond L, Favara C, Baron G, Priola S, et al. Anchorless prion protein results in infectious amyloid disease without clinical scrapie. Science 2005; 308:1435 - 9; http://dx.doi.org/10.1126/science.1110837; PMID: 15933194
- Chesebro B, Race B, Meade-White K, Lacasse R, Race R, Klingeborn M, Striebel J, Dorward D, McGovern G, Jeffrey M. Fatal transmissible amyloid encephalopathy: a new type of prion disease associated with lack of prion protein membrane anchoring. PLoS Pathog 2010; 6:e1000800; http://dx.doi.org/10.1371/journal.ppat.1000800; PMID: 20221436
- Stöhr J, Watts JC, Legname G, Oehler A, Lemus A, Nguyen H-OB, Sussman J, Wille H, DeArmond SJ, Prusiner SB, et al. Spontaneous generation of anchorless prions in transgenic mice. Proc Natl Acad Sci U S A 2011; 108:21223 - 8; http://dx.doi.org/10.1073/pnas.1117827108; PMID: 22160704
- Jansen C, Parchi P, Capellari S, Vermeij AJ, Corrado P, Baas F, Strammiello R, van Gool WA, van Swieten JC, Rozemuller AJM. Prion protein amyloidosis with divergent phenotype associated with two novel nonsense mutations in PRNP. Acta Neuropathol 2010; 119:189 - 97; http://dx.doi.org/10.1007/s00401-009-0609-x; PMID: 19911184
- Černilec M, Vranac T, Hafner-Bratkovic I, Koren S, Venturini AC, Popović M, Juntes P, Šerbec VČ. Identification of an epitope on the recombinant bovine PrP that is able to elicit a prominent immune response in wild-type mice. Immunol Lett 2007; 113:29 - 39; http://dx.doi.org/10.1016/j.imlet.2007.07.012; PMID: 17884181