
Abstract
Knockout of the cellular prion protein (PrPC) in mice is tolerated, as is complete elimination of the protein’s N-terminal domain. However, deletion of select short segments between the N- and C-terminal domains is lethal. How can one reconcile this apparent paradox? Research over the last few years demonstrates that PrPC undergoes α-cleavage in the vicinity of residue 109 (mouse sequence) to release the bioactive N1 and C1 fragments. In biophysical studies, we recently characterized the action of relevant members of the ADAM (A Disintegrin And Metalloproteinase) enzyme family (ADAM8, 10, and 17) and found that they all produce α-cleavage, but at 3 distinct cleavage sites, with proteolytic efficiency modulated by the physiologic metals copper and zinc. Remarkably, the shortest lethal deletion segment in PrPC fully encompasses the three α-cleavage sites. Analysis of all reported PrPC deletion mutants suggests that elimination of α-cleavage, coupled with retention of the protein’s N-terminal residues, segments 23–31 and longer, confers the lethal phenotype. Interestingly, these N-terminal residues are implicated in the activation of several membrane proteins, including synaptic glutamate receptors. We propose that α-cleavage is a general mechanism essential for downregulating PrPC’s intrinsic activity, and that blockage of proteolysis leads to constitutively active PrPC and consequent dyshomeostasis.
Introduction
To date, there is still no definitive function assigned to the prion protein (PrP). PrP has been implicated in transmembrane signaling,Citation1 metal ion redox regulation and trafficking,Citation2 cell adhesion,Citation3 neuron myelination,Citation4 and apoptosisCitation5 (see ref. Citation6 for a general review of PrP function). Several recent discoveries, however, provide new perspectives that may not only assign a more definitive function to PrP, but may also give new insight into the processes that lead to prion disease. Among these discoveries are the findings that PrP is enzymatically cleaved to produce bioactive fragments,Citation7,Citation8 and that full-length PrP directly interacts with a subfamily of glutamate receptors to regulate zinc transport.Citation9 This brief review will describe these new perspectives, the connection to PrP structural features, and put forth a hypothesis in which cleavage serves as an essential regulatory mechanism of PrP action.
PrPC Cleavage—the Standard Model
The full-length cellular prion protein, PrPC, is approximately 210 amino acids long and possesses a helical C-terminal domain and a partially structured N-terminal domain (). A notable feature of the N-terminal domain is the octarepeat segment (residues 59–90, mouse sequence) capable of binding physiologic zinc and copper.Citation10,Citation11 Between these 2 domains is a highly conserved hydrophobic segment often implicated in aggregation and the formation of PrP scrapie (PrPSc). presents the N- and C-terminal domains as spatially separated, however, recent structural work suggests an important tertiary contact promoted by zinc (see below). In addition to full-length PrPC, the protein is also found in several distinct truncated forms in vivo, with proteolytic products possessing unique bioactive properties.Citation7,Citation12 In healthy tissues, enzymatic cleavage of PrPC at approximately K109↓H110 (mouse sequence), termed α-cleavage, produces the N-terminal and C-terminal fragments, N1 and C1, respectively. The preponderance of recent evidence suggests that α-cleavage, which separates most of the PrP N-terminus from the folded C-terminus, is due to action from one or more members of the ADAM (A Disintegrin And Metalloproteinase) family of enzymes, specifically ADAM8, ADAM10, and ADAM17.Citation13,Citation14 α-Cleavage was originally assigned to ADAM10, but recent studies find that neither knocking out ADAM10 nor treatment with ADAM10 specific protease inhibitors are capable of blocking α-cleavage, and instead block proteolysis of PrPC near the C-terminal GPI anchor at G227↓R228. More recently, ADAM8 was identified as the protease responsible for α-cleavage in skeletal muscle tissue.Citation13
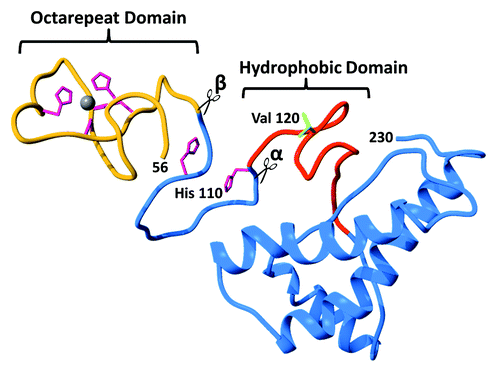
Figure 1. Ribbon diagram of the cellular prion protein (PrPC) showing the N-terminal octarepeat domain (gold), the folded C-terminal domain (blue), and the central hydrophobic region (red). Note that residues 23–55 are unstructured and omitted for clarity. The 4 His residues (magenta) in the octarepeat domain coordinate zinc and copper ions, as shown. The additional His residues at 95 and 110 are also capable of coordinating Cu2+. Previously proposed sites for α-cleavage β-cleavage are indicated at the beginning of the hydrophobic segment and the end of the octarepeat domain, respectively.
The pro-domains released by α-cleavage exhibit potent activities. The N1 fragment is antiapoptotic, possibly acting through the inhibition of caspase-3.Citation5 Conversely, the C1 fragment promotes apoptosis through p53 dependent caspase-3 activity, although it appears as though the protective effects of N1 significantly outweigh the pro-apoptotic effects of C1.Citation15 Perhaps more importantly, substoichiometric levels of C1 protect against PrPSc propagation.Citation16
PrPC also undergoes β-cleavage, which takes place at multiple sites within and just following the octarepeat domain, producing N2 and C2 fragments.Citation7 Experiments in CHO and SH-SY5Y cell lines expressing PrPC find that levels of C2 are greatly enhanced upon the addition of H2O2, suggesting proteolysis by reactive oxygen species (ROS) generated by intrinsic copper, a metal ion that binds with high affinity to the N-terminal octarepeats.Citation17 A separate pathway to β-cleavage of PrPSc is enzymatic, produced by calpainsCitation18 and cathepsinCitation19 proteases (see review ref. Citation20). In general, β-cleavage is observed in normal brain tissue, but C2 is enriched in prion infection. Unlike the N1 and C1 fragments, N2 and C2 do not show any bioactivity or neuroprotection, although β-cleavage’s production of N2 and C2 may indirectly assert a biological effect by prohibiting the formation of N1.
Zinc Promoted Tertiary Contacts in PrPC
The prion protein’s 2 domains are often thought to be noninteracting—a structured C terminus with an N-terminal tail. Cellular and biophysical studies, however, suggest that the 2 domains influence each other in a way that is relevant to both PrPC function and misfolding in disease.Citation21 It is now well established that physiologic concentrations of copper (Cu2+) and zinc (Zn2+) promote coordination of these metal ions to the octarepeat domain and, in the case of Cu2+, to 2 nearby sites, as well.Citation10,Citation11,Citation22 With the goal of trying to develop concepts for PrPC function, our lab applied electron paramagnetic resonance (EPR) to evaluate the Cu2+ coordination features. At low copper concentration, the Cu2+ ion binds with high affinity (~0.10 nM dissociation constant) to the 4 histidine imidazoles in the octarepeat domain.Citation2,Citation23 Using peptide mapping, we found a similar coordination sphere for Zn2+, although with a weaker dissociation constant of ~200 µM.Citation24
Given the potential for N-terminal—C-terminal interactions, and the fact that previous structural studies did not include metal ions, we investigated whether Zn2+ might promote higher order structure in PrPC. NMR shows that the Zn2+ occupied octarepeat domain folds back on helices 2 and 3 of the C-terminal domain forming a previously unseen tertiary contact.Citation25 This newly identified contact is further supported by direct interdomain distance measurements using double electron-electron resonance (DEER) EPR. Our work parallels NMR studies with Cu2+ that also find a metal ion promoted interaction.Citation26 An energy-minimized model developed with constraints from our NMR and EPR experiments is shown in . Interestingly, helices 2 and 3 possess the majority of the point mutations that cause familial prion disease, and NMR data suggest that these mutations weaken the interdomain interaction.Citation25
Figure 2. Global PrPC fold promoted by Zn2+, as demonstrated by Spevacek et al. using NMR and double label electron paramagnetic resonance.Citation25 The Zn2+ occupied octarepeat domain makes a tertiary contact with the surface formed by C-terminal helices 2 and 3. Note that helices 2 and 3 present a cluster of negatively charged residues that facilitate the tertiary contact, and that familial mutations weaken the fold.
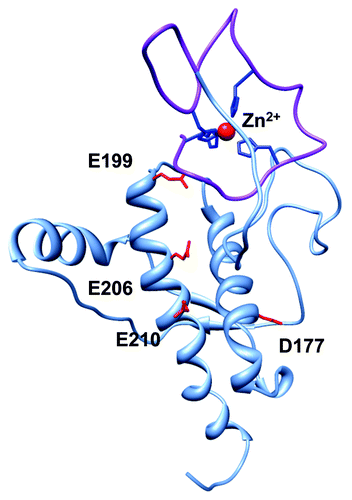
Early biophysical investigations into the thermodynamic basis for inherited prion disease hypothesized that mutations would thermodynamically destabilize the C-terminal structure of PrPC rendering the protein susceptible to misfolding. However, structure and denaturation studies reveal only minimal changes in three-dimensional fold, and no systematic variation in stability.Citation27,Citation28 Our investigations find that pathogenic mutants, such as E199K (mouse sequence; E200K in human), systematically alter the N-terminal–C-terminal tertiary interaction, likely by reducing the negative surface charge potential on helices 2 and 3 that attracts the Zn2+-bound N-terminus.Citation25 Thus, this class of charge altering mutations may reduce stability by affecting the metal ion promoted tertiary fold in PrPC. Conversely, in wild type PrP perhaps the metal promoted interaction explains why both copper and zinc inhibit in vitro PrPres amplification.Citation29
PrPC Function
Global perspectives of PrP function have been thoroughly reviewed elsewhere (see Aguzzi et al. for example, ref. Citation6). With regard to metal ions, it is now well established that both copper and zinc stimulate PrPC internalization through endocytosisCitation30 and that copper activates transcription factors leading to an increase of PrPC expression.Citation31,Citation32 In general, PrPC appears to be important in copper/zinc homeostasis and serves to protect cellular components when elevated metal ion levels would otherwise lead to cellular stress. Based on an analysis of coordination features, we’ve proposed that PrPC acts to suppress copper electrochemistry that, if left unregulated, could lead to the production of free radicals and reactive oxygen species.Citation2 We also found that expansion of the octarepeat domain leads to altered copper uptake in a way that suggests a strong relationship to inherited prion disease.Citation33 Recent electrochemical studies lend support to this hypothesis and show further that the PrPC-copper complex produces low levels of hydrogen peroxide, which may participate in cellular signaling.Citation34,Citation35
Watt et al. identified a fundamentally new function for PrPC in which the protein participates in neuronal zinc uptake. Using selective dyes, they showed that PrPC enhances zinc uptake through AMPA receptors, a member of the multi-subunit glutamate receptor family.Citation36 PrPC and AMPA co-precipitate, demonstrating a physical interaction between the 2 membrane proteins. Both the PrP octarepeat domain and the polybasic N-terminus, corresponding to residues 23–26 (Lys-Lys-Arg-Pro; mouse), were required for zinc transport, but only the polybasic region was required for co-precipitation. Watt et al. suggest that PrPC provides neuroprotection by facilitating zinc reuptake following glutamate release, thereby regulating synaptic zinc levels. In this scenario, PrPC docks to AMPA through its N-terminus and the octarepeat domain loads Zn2+ in preparation for transport through the AMPA channel.Citation9 Interestingly, 2 familial mutants—P101L and D177N—shown by Spevacek et al. to weaken the Zn2+ promoted tertiary structure also lower zinc transport, likely by disrupting the PrPC–AMPA physical interaction.
PrPC Cleavage Revisited
As outlined above, the prevailing paradigm of PrPC cleavage posits that α-cleavage is enzymatically driven and constitutes normal processing, while β-cleavage results from aberrant copper redox activity and is associated with the development of prion disease. But the identification of several ADAM family enzymes producing α-cleavage, along with the structural features promoted by copper and zinc, motivated a reassessment of PrPC proteolysis. We used liquid chromatography/mass spectrometry (LC/MS) to analyze the rates and fragmentation patterns of PrPC resulting from the redox activity of copper, and proteolysis from ADAM8, ADAM10, and ADAM17.Citation8 Enzymes were assayed in the absence and presence of Cu2+ and Zn2+.
shows the identified proteolysis sites. all three enzymes produce α-cleavage, but at distinct locations, which we refer to as α1 (following residue 109; ADAM8 primary site), α2 (following residue 116; ADAM8 minor site), and α3 (following residue 119; ADAM10 and ADAM17). ADAM10 also functions as a sheddase, cleaving PrPC near the C-terminus to release the protein from the membrane surface.Citation37 Under reducing conditions, produced by the addition of ascorbic acid, copper alone produces extensive β-cleavage, consistent with prevailing hypotheses. Unexpectedly, ADAM8 also produces extensive β-cleavage with multiple proteolysis sites throughout the octarepeat domain. Consequently, β-cleavage may result from both copper redox activity and ADAM8 proteolysis.
Figure 3. Summary of the findings of McDonald et al. on the α-cleavage and β-cleavage by ADAM family enzymes.Citation8 N-terminal (white) and C-terminal (green) domains are shown, with octarepeats (gold), as is the C-terminal GPI moiety that anchors the protein to the extracellular membrane leaflet. (A) ADAM8, ADAM10, and ADAM17 produce α-cleavage at distinct sites, noted as α1, α2, and α3. ADAM10 also acts as a sheddase by cleaving near the C-terminus to release the protein from the membrane. Also shown are the sites in the octarepeat domain where ADAM8 produces β-cleavage. (B) Relative cleavage activities are indicated by arrow thickness. ADAM8 alone (no metal ions) produces extensive α1- and β-cleavage, with α2-cleavage as a minor product. (C) In the presence of Cu2+, α1- and β-cleavage are suppressed, while α2-cleavage is enhanced. (D) Zn2+ also inhibits β-cleavage but, in contrast to Cu2+, promotes α1- over α2-cleavage. In contrast to the previous paradigm, these findings show that metal ions suppress β-cleavage and template a structure that promotes α-cleavage.
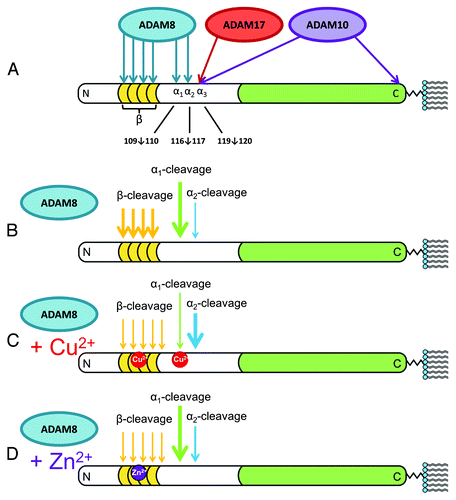
Zinc is inert with respect to redox activity, and so is copper in the absence of the ascorbic acid reductant, but both metal ions strongly influence the proteolytic susceptibility to ADAM8, the primary enzyme in muscle responsible for α-cleavage. As shown in , Cu2+ inhibits β-cleavage and promotes α2-cleavage while Zn2+ similarly inhibits β-cleavage but promotes α1-cleavage. Interestingly, elimination of the octarepeat domain in parallel experiments with Cu2+ fails to find an enhancement of α2-cleavage suggesting that the metal ion promoted tertiary fold in PrPC facilitates specific cleavage patterns. Together, our results find that α-cleavage takes place at several nearby sites between residues 109 and 120, and that metal ions actually suppress β-cleavage, while modulating the α1/α2 product ratios.
Emphasizing the importance of α-cleavage to release the N1 and C1 PrPC fragments, Oliveira-Martins et al. examined PrPC proteolysis in a wide range of cell lines.Citation38 Production of the N1/C1 fragments was universal, thereby underscoring the physiological importance of α-cleavage. It was thought at the time that cleavage was localized exclusively to the 109↓110 position, yet replacing the residues flanking this site only lowered proteolytic yields, but did not fully eliminate it. In general, mutations producing changes in local charge or hydrophobicity exerted only a modest reduction in α-cleavage efficiency. In contrast, polypeptide deletions were effective and removal of progressively larger stretches within the segment 106–119 produced a linear reduction in cleavage, with complete deletion of this segment required to fully suppress proteolysis. The authors proposed that perhaps several PrPase enzymes might be active in this region. Following our characterization of PrPC proteolysis, one would need to eliminate the α1, α2, and α3 sites, in the segment 109–120, to fully eliminate α-cleavage. Perhaps the physiologic requirement for α-cleavage is sufficiently important to be preserved through the evolution of redundant mechanisms.
PrPC Deletions and Toxicity
Complete elimination of the prion protein in transgenic mice is well tolerated, yet paradoxically, deletion of certain N-terminal PrP segments can be lethal. Schmerling et al. explored a series of N-terminal deletions starting at residue 32, and extending progressively toward the C-terminal domain, and found that those ending at residue 121 or beyond produce ataxia, cerebellar degeneration, and death after several months.Citation39 Interestingly, as shown by Baumann et al., retaining more of the PrPC N-terminus (Δ94–134) leads to an even more toxic phenotype, with myelin degeneration and death within 20–30 d.Citation4 Consistent with this trend, Li et al. found that mice expressing PrPC(Δ105–125)—a deletion of only 21 residues—develop severe neurodegenerative symptoms leading to lethality approximately a week after birth.Citation40 In all cases, PrPC deletion toxicity was rescued by coexpression with wild type PrPC, with the more toxic mutants requiring higher relative levels of wild type protein.
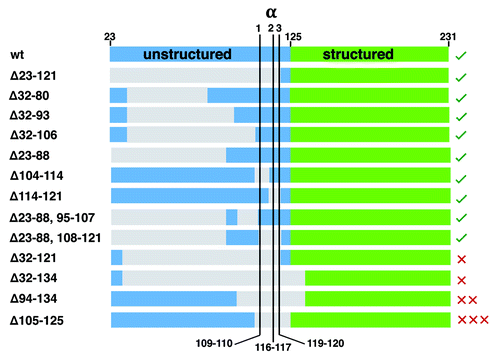
We collected the specific sequences from studies of mouse PrPC deletion mutants, focusing on those with the N-terminal signal peptide (residues 1–22) and the C-terminal GPI signal sequence (to ensure export and attachment to the extracellular membrane), and show them aligned in . Sequences are ordered with benign deletion mutants at the top, moving to progressively more toxic phenotypes at the bottom. Sites for the recently characterized α1, α2, and α3-cleavage are shown. There are 2 complementary patterns. First, deletions that fully remove all three α-cleavage sites are toxic, but only if N-terminal residues 23–31 are retained. Second, with deletion of the α-cleavage sites, the longer the retained N-terminal segment, the greater the toxicity, as measured by the copy number of wild type PrPC required for rescue. Highlighting this pattern, PrPC(Δ23–121) is not toxic, whereas PrPC(Δ105–125) gives the most lethal phenotype. It’s interesting to compare 2 tolerated deletion proteins PrPC(Δ104–114) and PrPC(Δ114–121). While each possesses most of the N-terminus, but with partial elimination of the α-cleavage sites, they retain the α2 and α1 sites, respectively, and thus remain susceptible to ADAM8 proteolysis.
Figure 4. Summary of PrPC deletion mutants and resulting phenotypes (sequence details summarized in Aguzzi et al. ref. Citation6). The N-terminal domain after removal of the signal peptide starts at residue 23, and is shown in blue, while the structured C-terminal domain, starting at approximately residue 125 is shown in green. Deleted segments, as noted by the numbering (left), are gray. Locations for α1, α2, and α3-cleavage are shown. The green checks, right, indicate tolerated deletion mutants, whereas the red X’s are the lethal mutants. The number of Xs qualitatively represents the lethal potency of each mutant, as indicated by the relative amount of wildtype PrPC required for rescue. This alignment shows that deletion of the full α-cleavage segment, 109–120, but with retention of N-terminal residues starting at position 23 confers toxicity.
PrPC Overdrive—Toxic Deletions Eliminate α-Cleavage
Deletion studies have prompted several different hypotheses, with suggestions of a PrPC binding partner where interaction is facilitated by a segment in the vicinity of 105–125. Either the segment alone is essential for bindingCitation40 or, alternatively, that PrPC binds in a bivalent fashion through its N- and C-terminal domains with a region encompassing 105–125 serving as a linker.Citation4,Citation39 Another line of inquiry suggests that PrPC(Δ105–125) facilitates membrane leakiness by forming spontaneous ion channels, an effect that is blocked by interaction with wildtype PrPC.Citation41
The recent appreciation of the physiologic importance of proteolysis leading to N1 and C1, along with redundant cleavage sites and enzymes targeted to 109–120, suggest a different scenario. We propose that full-length PrPC modulates the activity of one or more nearby companion membrane proteins, such as the AMPA receptor, and that this activity is downregulated by α-cleavage. Any deletion that fully interrupts α-cleavage leads to constitutive activity of PrPC and its binding partner, which promotes the toxic response and subsequent neurodegeneration. For example, in the case of the AMPA receptor, neurons may hyper accumulate zinc. Similarly, any molecular species that blocks protease access to the α-cleavage sites will also produce a toxic response.
provides a schematic using zinc transport through the AMPA receptor as an example. Considering the previous sections, we note that (1) the PrPC N-terminus makes direct contact with the receptor, (2) the octarepeat domain is required for enhanced zinc transport through the AMPA receptor, and (3) that copper and zinc promote α-cleavage. As shown stepwise in , and in accord with a model previously advanced by Watt et al.,Citation9 Zn2+ binds to the PrPC octarepeat domain, which in turn, drives the compact fold identified by Spevacek et al.Citation25 PrPC then associates with the AMPA receptor, thorough its N-terminal segment, leading to enhanced Zn2+ transport. Zn2+ structures the segment 109–120 so that it is more susceptible to ADAM cleavage, which turns off PrPC enhanced AMPA receptor activity. PrPC fragments (N1 and C1) then dissociate from the AMPA receptor. The net effect of this mechanism is that PrPC facilitates the tightly controlled trafficking of Zn2+ into the cell. Interestingly, an evolutionary cousin of PrPC, the zinc transporter ZIP10 also employs a Zn2+ mediated interdomain interaction, as well as Zn2+ dependent cleavage of the N-terminus to modulate the ability of ZIP10 to uptake zinc.Citation42 We note that with PrPC, the specific enzyme responsible for α-cleavage is certain to be tissue dependent, and there is yet no evidence of significant ADAM8 activity in the brain.
Figure 5. A proposal for PrPC regulation through α-cleavage. (A) The AMPA receptor (AMPAR), a cation channel, is one of several membrane proteins thought to be modulated by PrPC binding. The folded PrPC C-terminal domain (square) is GPI linked to the extracellular membrane. The N-terminal domain is unstructured in the absence of Zn2+. (B) Zn2+ binds to the octarepeats, structuring the N-terminal domain, which then associates with a negatively charged patch on the C-terminal domain. Residues 23 – 31 bind to the AMPAR facilitating Zn2+ transport. The PrPC fold promoted by Zn2+ exposes the α-cleavage site, which becomes susceptible to the ADAM proteases (8, 10, and 17). (C) α-Cleavage releases N1, thus downregulating PrPC facilitated Zn2+ transport. C-Terminal PrPC (the C1 fragment) is degraded and replaced by full-length PrPC. (D) Occlusion of α-cleavage (skull and crossbones) by the peptide PrP(106–126) or Aβo, for example, or by misfolding to PrPSc, leaves PrP in a constitutively active state, thus driving dyshomeostasis.
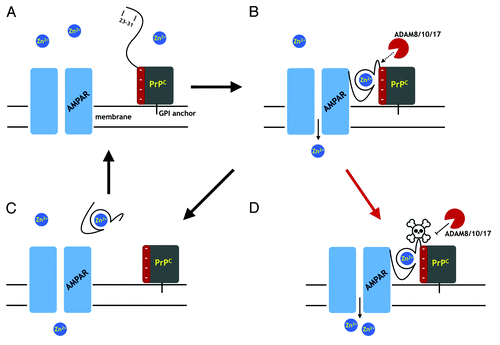
A consequence of the model in is that interfering with access to the α-cleavage would lead to persistent and elevated Zn2+ transport, perhaps disrupting cellular homeostasis. A partial test of toxicity arising from proteolytically resistant PrPC would be select point mutations, as opposed to deletions, designed to suppress α-cleavage with minimal alteration of the native protein sequence or structure. We recently developed the mutants PrPC(H110Y) and PrPC(V120D), which block α1- and α3-cleavage, respectively, thereby offering strategies for disentangling the toxic responses from different cleavage patterns.Citation8
The scheme presented in offers mechanisms for how coexpression of wild type PrPC rescues the syndrome resulting from deletion of the proteolytic segment. Release of the wild type PrPC N1 peptide, which alone is unlikely to facilitate zinc transport, could compete with mutant PrPC for the AMPA receptor binding site. Similarly, full-length or C1 wild type PrPC could associate with the AMPA receptor driving off the mutant protein.
Could this model, which posits the essential need for PrPC inactivation by α-cleavage, help explain other experiments demonstrating neurotoxic effects and perhaps certain aspects of neurodegenerative disease? The short peptide PrP(106–126) is toxic when dosed into cultured neurons, but only if wild type PrP is present.Citation43 This particular stretch of PrP is hydrophobic and amyloidogenic and, consistent with fibril forming peptides, forms self-complementary ultra structures. Thus, it likely targets the PrPC segment 106–126, which perfectly overlaps the α-cleavage region thereby reducing susceptibility to proteolysis. In prion disease, the prion protein misfolds and assembles into aggregates forming PrPSc, which is partially protease resistant. Experimental treatment of purified PrPSc with proteinase K cleaves at approximately residue 90,Citation44 suggesting that the N-terminal segment is largely exposed but that the α-cleavage region beyond residue 90 is occluded. As with PrP(106–126) this would inhibit normal proteolysis needed to inactivate PrP’s influence on the AMPA receptor. Recent work in Alzheimer disease finds that the most toxic species formed by Aβ aggregates are oligomers, often noted as Aβo.Citation45 In hippocampal slices Aβo inhibits long-term potentiation, but only in the presence of PrPC; brain slices derived from PrP knockouts are resistant to Aβo toxicity.Citation46 Fluharty et al. have demonstrated direct binding between Aβo and PrPC and mapped the interaction to 2 N-terminal regions, residues 23–27 and 95–105.Citation47 The segment 95–105 is just on the N-terminal side of the α1-cleavage site and Aβo binding would likely block access for proteolysis. Additionally, Aβo sequesters N1, removing the ability of N1 to regulate PrPC activation of membrane receptors. With regard to the specific activation of the AMPA receptor, emerging theories of Alzheimer disease point to the importance of maintaining zinc homeostasis, the breakdown of which promotes the formation of neurofibrillary tangles.Citation48
Much of our discussion above has centered on PrPC activation of zinc transport through the AMPA receptor. However, PrPC may have other membrane targets. For example, Um et al. find that PrPC-Aβo couples to Fyn kinase activation through the metabotropic glutamate receptor 5 (mGluR5).Citation45 At this juncture, it is not clear whether PrPC binds to mGluR5 in the absence of Aβo. PrPC also modulates currents through the NMDA receptor in a copper-dependent fashion.Citation49 Specifically, copper occupied PrPC reduces the receptor’s sensitivity to glycine, which promotes persistent cationic currents, an effect that is tempered by both monomeric Aβ and Aβo. PrPC is trafficked by endocytosis through interaction with the LDL receptor-related protein 1 (LRP1), identifying yet another membrane partner. Taken together, PrPC may have multiple interaction partners and the scenario we developed for inactivation may be a universal regulatory mechanism.
We have emphasized constitutive PrPC activity through elimination of the α-cleavage sites as a primary mechanism for driving dyshomeostasis. This concept supports the notion of an executive function for the PrPC N-terminal domain, where interference between the protein’s 2 intact domains enhances toxicity.Citation50 However, there is ample evidence that N1, in particular, is an important signaling species.Citation5 Without α-cleavage, N1 production is eliminated, which could facilitate apoptosis. If this mechanism alone were responsible for the neurodegeneration resulting from toxic PrPC deletion mutants, it would be difficult to understand the relatively mild phenotype resulting from PrPC knockout.
Prion disease and Alzheimer disease are marked by a wide set of neurological derangements and it is not clear whether constitutive activity driven by hindered proteolysis of PrPC is a major component. However, brain zinc levels are lowered in prion disease suggesting altered PrPC function. In general, there are ongoing discussions as to whether neurodegenerative disease arises from a loss of normal protein function or a gain of function from toxic aggregates. We argue here for a third possibility where intrinsic PrPC function is constitutively maintained thereby leading to neuronal stress. If correct, this scheme could lead to new avenues for treating neurodegenerative processes.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
The authors thank Eric G.B. Evans for suggestions and editorial comments on the manuscript. This work was supported by a grant from the National Institutes of Health (GM065790).
Related Research Data
References
- Mouillet-Richard S, Ermonval M, Chebassier C, Laplanche JL, Lehmann S, Launay JM, Kellermann O. Signal transduction through prion protein. Science 2000; 289:1925 - 8; http://dx.doi.org/10.1126/science.289.5486.1925; PMID: 10988071
- Chattopadhyay M, Walter ED, Newell DJ, Jackson PJ, Aronoff-Spencer E, Peisach J, Gerfen GJ, Bennett B, Antholine WE, Millhauser GL. The octarepeat domain of the prion protein binds Cu(II) with three distinct coordination modes at pH 7.4. J Am Chem Soc 2005; 127:12647 - 56; http://dx.doi.org/10.1021/ja053254z; PMID: 16144413
- Schmitt-Ulms G, Legname G, Baldwin MA, Ball HL, Bradon N, Bosque PJ, Crossin KL, Edelman GM, DeArmond SJ, Cohen FE, et al. Binding of neural cell adhesion molecules (N-CAMs) to the cellular prion protein. J Mol Biol 2001; 314:1209 - 25; http://dx.doi.org/10.1006/jmbi.2000.5183; PMID: 11743735
- Baumann F, Tolnay M, Brabeck C, Pahnke J, Kloz U, Niemann HH, Heikenwalder M, Rülicke T, Bürkle A, Aguzzi A. Lethal recessive myelin toxicity of prion protein lacking its central domain. EMBO J 2007; 26:538 - 47; http://dx.doi.org/10.1038/sj.emboj.7601510; PMID: 17245436
- Guillot-Sestier MV, Sunyach C, Druon C, Scarzello S, Checler F. The alpha-secretase-derived N-terminal product of cellular prion, N1, displays neuroprotective function in vitro and in vivo. J Biol Chem 2009; 284:35973 - 86; http://dx.doi.org/10.1074/jbc.M109.051086; PMID: 19850936
- Aguzzi A, Baumann F, Bremer J. The prion’s elusive reason for being. Annu Rev Neurosci 2008; 31:439 - 77; http://dx.doi.org/10.1146/annurev.neuro.31.060407.125620; PMID: 18558863
- Chen SG, Teplow DB, Parchi P, Teller JK, Gambetti P, Autilio-Gambetti L. Truncated forms of the human prion protein in normal brain and in prion diseases. J Biol Chem 1995; 270:19173 - 80; http://dx.doi.org/10.1074/jbc.270.32.19173; PMID: 7642585
- McDonald AJ, Dibble JP, Evans EG, Millhauser GL. A new paradigm for enzymatic control of α-cleavage and β-cleavage of the prion protein. J Biol Chem 2014; 289:803 - 13; http://dx.doi.org/10.1074/jbc.M113.502351; PMID: 24247244
- Watt NT, Griffiths HH, Hooper NM. Neuronal zinc regulation and the prion protein. Prion 2013; 7:203 - 8; http://dx.doi.org/10.4161/pri.24503; PMID: 23764834
- Millhauser GL. Copper binding in the prion protein. Acc Chem Res 2004; 37:79 - 85; http://dx.doi.org/10.1021/ar0301678; PMID: 14967054
- Millhauser GL. Copper and the prion protein: methods, structures, function, and disease. Annu Rev Phys Chem 2007; 58:299 - 320; http://dx.doi.org/10.1146/annurev.physchem.58.032806.104657; PMID: 17076634
- Guillot-Sestier MV, Checler F. α-Secretase-derived cleavage of cellular prion yields biologically active catabolites with distinct functions. Neurodegener Dis 2012; 10:294 - 7; http://dx.doi.org/10.1159/000333804; PMID: 22261541
- Liang J, Wang W, Sorensen D, Medina S, Ilchenko S, Kiselar J, Surewicz WK, Booth SA, Kong Q. Cellular prion protein regulates its own α-cleavage through ADAM8 in skeletal muscle. J Biol Chem 2012; 287:16510 - 20; http://dx.doi.org/10.1074/jbc.M112.360891; PMID: 22447932
- Vincent B, Paitel E, Saftig P, Frobert Y, Hartmann D, De Strooper B, Grassi J, Lopez-Perez E, Checler F. The disintegrins ADAM10 and TACE contribute to the constitutive and phorbol ester-regulated normal cleavage of the cellular prion protein. J Biol Chem 2001; 276:37743 - 6; PMID: 11477090
- Sunyach C, Cisse MA, da Costa CA, Vincent B, Checler F. The C-terminal products of cellular prion protein processing, C1 and C2, exert distinct influence on p53-dependent staurosporine-induced caspase-3 activation. J Biol Chem 2007; 282:1956 - 63; http://dx.doi.org/10.1074/jbc.M609663200; PMID: 17121821
- Westergard L, Turnbaugh JA, Harris DA. A naturally occurring C-terminal fragment of the prion protein (PrP) delays disease and acts as a dominant-negative inhibitor of PrPSc formation. J Biol Chem 2011; 286:44234 - 42; http://dx.doi.org/10.1074/jbc.M111.286195; PMID: 22025612
- Watt NT, Hooper NM. Reactive oxygen species (ROS)-mediated beta-cleavage of the prion protein in the mechanism of the cellular response to oxidative stress. Biochem Soc Trans 2005; 33:1123 - 5; http://dx.doi.org/10.1042/BST20051123; PMID: 16246061
- Yadavalli R, Guttmann RP, Seward T, Centers AP, Williamson RA, Telling GC. Calpain-dependent endoproteolytic cleavage of PrPSc modulates scrapie prion propagation. J Biol Chem 2004; 279:21948 - 56; http://dx.doi.org/10.1074/jbc.M400793200; PMID: 15026410
- Dron M, Moudjou M, Chapuis J, Salamat MK, Bernard J, Cronier S, Langevin C, Laude H. Endogenous proteolytic cleavage of disease-associated prion protein to produce C2 fragments is strongly cell- and tissue-dependent. J Biol Chem 2010; 285:10252 - 64; http://dx.doi.org/10.1074/jbc.M109.083857; PMID: 20154089
- Altmeppen HC, Puig B, Dohler F, Thurm DK, Falker C, Krasemann S, Glatzel M. Proteolytic processing of the prion protein in health and disease. Am J Neurodegener Dis 2012; 1:15 - 31; PMID: 23383379
- Li L, Guest W, Huang A, Plotkin SS, Cashman NR. Immunological mimicry of PrPC-PrPSc interactions: antibody-induced PrP misfolding. Protein Eng Des Sel 2009; 22:523 - 9; http://dx.doi.org/10.1093/protein/gzp038; PMID: 19602568
- Walter ED, Stevens DJ, Spevacek AR, Visconte MP, Dei Rossi A, Millhauser GL. Copper binding extrinsic to the octarepeat region in the prion protein. Curr Protein Pept Sci 2009; 10:529 - 35; http://dx.doi.org/10.2174/138920309789352056; PMID: 19538144
- Walter ED, Chattopadhyay M, Millhauser GL. The affinity of copper binding to the prion protein octarepeat domain: evidence for negative cooperativity. Biochemistry 2006; 45:13083 - 92; http://dx.doi.org/10.1021/bi060948r; PMID: 17059225
- Walter ED, Stevens DJ, Visconte MP, Millhauser GL. The prion protein is a combined zinc and copper binding protein: Zn2+ alters the distribution of Cu2+ coordination modes. J Am Chem Soc 2007; 129:15440 - 1; http://dx.doi.org/10.1021/ja077146j; PMID: 18034490
- Spevacek AR, Evans EG, Miller JL, Meyer HC, Pelton JG, Millhauser GL. Zinc drives a tertiary fold in the prion protein with familial disease mutation sites at the interface. Structure 2013; 21:236 - 46; http://dx.doi.org/10.1016/j.str.2012.12.002; PMID: 23290724
- Thakur AK, Srivastava AK, Srinivas V, Chary KV, Rao CM. Copper alters aggregation behavior of prion protein and induces novel interactions between its N- and C-terminal regions. J Biol Chem 2011; 286:38533 - 45; http://dx.doi.org/10.1074/jbc.M111.265645; PMID: 21900252
- Liemann S, Glockshuber R. Influence of amino acid substitutions related to inherited human prion diseases on the thermodynamic stability of the cellular prion protein. Biochemistry 1999; 38:3258 - 67; http://dx.doi.org/10.1021/bi982714g; PMID: 10079068
- Bae SH, Legname G, Serban A, Prusiner SB, Wright PE, Dyson HJ. Prion proteins with pathogenic and protective mutations show similar structure and dynamics. Biochemistry 2009; 48:8120 - 8; http://dx.doi.org/10.1021/bi900923b; PMID: 19618915
- Orem NR, Geoghegan JC, Deleault NR, Kascsak R, Supattapone S. Copper (II) ions potently inhibit purified PrPres amplification. J Neurochem 2006; 96:1409 - 15; http://dx.doi.org/10.1111/j.1471-4159.2006.03650.x; PMID: 16417569
- Pauly PC, Harris DA. Copper stimulates endocytosis of the prion protein. J Biol Chem 1998; 273:33107 - 10; http://dx.doi.org/10.1074/jbc.273.50.33107; PMID: 9837873
- Bellingham SA, Coleman LA, Masters CL, Camakaris J, Hill AF. Regulation of prion gene expression by transcription factors SP1 and metal transcription factor-1. J Biol Chem 2009; 284:1291 - 301; http://dx.doi.org/10.1074/jbc.M804755200; PMID: 18990686
- Qin K, Zhao L, Ash RD, McDonough WF, Zhao RY. ATM-mediated transcriptional elevation of prion in response to copper-induced oxidative stress. J Biol Chem 2009; 284:4582 - 93; http://dx.doi.org/10.1074/jbc.M808410200; PMID: 19064990
- Stevens DJ, Walter ED, Rodríguez A, Draper D, Davies P, Brown DR, Millhauser GL. Early onset prion disease from octarepeat expansion correlates with copper binding properties. PLoS Pathog 2009; 5:e1000390; http://dx.doi.org/10.1371/journal.ppat.1000390; PMID: 19381258
- Liu L, Jiang D, McDonald A, Hao Y, Millhauser GL, Zhou F. Copper redox cycling in the prion protein depends critically on binding mode. J Am Chem Soc 2011; 133:12229 - 37; http://dx.doi.org/10.1021/ja2045259; PMID: 21707094
- Zhou F, Millhauser GL. The Rich Electrochemistry and Redox Reactions of the Copper Sites in the Cellular Prion Protein. Coord Chem Rev 2012; 256:2285 - 96; http://dx.doi.org/10.1016/j.ccr.2012.04.035; PMID: 23144499
- Watt NT, Taylor DR, Kerrigan TL, Griffiths HH, Rushworth JV, Whitehouse IJ, Hooper NM. Prion protein facilitates uptake of zinc into neuronal cells. Nat Commun 2012; 3:1134; http://dx.doi.org/10.1038/ncomms2135; PMID: 23072804
- Taylor DR, Parkin ET, Cocklin SL, Ault JR, Ashcroft AE, Turner AJ, Hooper NM. Role of ADAMs in the ectodomain shedding and conformational conversion of the prion protein. J Biol Chem 2009; 284:22590 - 600; http://dx.doi.org/10.1074/jbc.M109.032599; PMID: 19564338
- Oliveira-Martins JB, Yusa S, Calella AM, Bridel C, Baumann F, Dametto P, Aguzzi A. Unexpected tolerance of alpha-cleavage of the prion protein to sequence variations. PLoS One 2010; 5:e9107; http://dx.doi.org/10.1371/journal.pone.0009107; PMID: 20161712
- Shmerling D, Hegyi I, Fischer M, Blättler T, Brandner S, Götz J, Rülicke T, Flechsig E, Cozzio A, von Mering C, et al. Expression of amino-terminally truncated PrP in the mouse leading to ataxia and specific cerebellar lesions. Cell 1998; 93:203 - 14; http://dx.doi.org/10.1016/S0092-8674(00)81572-X; PMID: 9568713
- Li A, Christensen HM, Stewart LR, Roth KA, Chiesa R, Harris DA. Neonatal lethality in transgenic mice expressing prion protein with a deletion of residues 105-125. EMBO J 2007; 26:548 - 58; http://dx.doi.org/10.1038/sj.emboj.7601507; PMID: 17245437
- Biasini E, Turnbaugh JA, Unterberger U, Harris DA. Prion protein at the crossroads of physiology and disease. Trends Neurosci 2012; 35:92 - 103; http://dx.doi.org/10.1016/j.tins.2011.10.002; PMID: 22137337
- Ehsani S, Salehzadeh A, Huo H, Reginold W, Pocanschi CL, Ren H, Wang H, So K, Sato C, Mehrabian M, et al. LIV-1 ZIP ectodomain shedding in prion-infected mice resembles cellular response to transition metal starvation. J Mol Biol 2012; 422:556 - 74; http://dx.doi.org/10.1016/j.jmb.2012.06.003; PMID: 22687393
- Forloni G, Angeretti N, Chiesa R, Monzani E, Salmona M, Bugiani O, Tagliavini F. Neurotoxicity of a prion protein fragment. Nature 1993; 362:543 - 6; http://dx.doi.org/10.1038/362543a0; PMID: 8464494
- Prusiner SB, Groth DF, Bolton DC, Kent SB, Hood LE. Purification and structural studies of a major scrapie prion protein. Cell 1984; 38:127 - 34; http://dx.doi.org/10.1016/0092-8674(84)90533-6; PMID: 6432339
- Um JW, Strittmatter SM. Amyloid-β induced signaling by cellular prion protein and Fyn kinase in Alzheimer disease. Prion 2013; 7:37 - 41; http://dx.doi.org/10.4161/pri.22212; PMID: 22987042
- Laurén J, Gimbel DA, Nygaard HB, Gilbert JW, Strittmatter SM. Cellular prion protein mediates impairment of synaptic plasticity by amyloid-beta oligomers. Nature 2009; 457:1128 - 32; http://dx.doi.org/10.1038/nature07761; PMID: 19242475
- Fluharty BR, Biasini E, Stravalaci M, Sclip A, Diomede L, Balducci C, La Vitola P, Messa M, Colombo L, Forloni G, et al. An N-terminal fragment of the prion protein binds to amyloid-β oligomers and inhibits their neurotoxicity in vivo. J Biol Chem 2013; 288:7857 - 66; http://dx.doi.org/10.1074/jbc.M112.423954; PMID: 23362282
- Craddock TJ, Tuszynski JA, Chopra D, Casey N, Goldstein LE, Hameroff SR, Tanzi RE. The zinc dyshomeostasis hypothesis of Alzheimer’s disease. PLoS One 2012; 7:e33552; http://dx.doi.org/10.1371/journal.pone.0033552; PMID: 22457776
- Stys PK, You H, Zamponi GW. Copper-dependent regulation of NMDA receptors by cellular prion protein: implications for neurodegenerative disorders. J Physiol 2012; 590:1357 - 68; http://dx.doi.org/10.1113/jphysiol.2011.225276; PMID: 22310309
- Sonati T, Reimann RR, Falsig J, Baral PK, O’Connor T, Hornemann S, Yaganoglu S, Li B, Herrmann US, Wieland B, et al. The toxicity of antiprion antibodies is mediated by the flexible tail of the prion protein. Nature 2013; 501:102 - 6; http://dx.doi.org/10.1038/nature12402; PMID: 23903654