
Abstract
The conformational conversion of the cellular prion protein (PrPC) to the β-rich infectious isoform PrPSc is considered a critical and central feature in prion pathology. Although PrPSc is the critical component of the infectious agent, as proposed in the “protein-only” prion hypothesis, cellular components have been identified as important cofactors in triggering and enhancing the conversion of PrPC to proteinase K resistant PrPSc. A number of in vitro systems using various chemical and/or physical agents such as guanidine hydrochloride, urea, SDS, high temperature, and low pH, have been developed that cause PrPC conversion, their amplification, and amyloid fibril formation often under non-physiological conditions. In our ongoing efforts to look for endogenous and exogenous chemical mediators that might initiate, influence, or result in the natural conversion of PrPC to PrPSc, we discovered that lipopolysaccharide (LPS), a component of gram-negative bacterial membranes interacts with recombinant prion proteins and induces conversion to an isoform richer in β sheet at near physiological conditions as long as the LPS concentration remains above the critical micelle concentration (CMC). More significant was the LPS mediated conversion that was observed even at sub-molar ratios of LPS to recombinant ShPrP (90–232).
Introduction
Prion diseases or transmissible spongiform encephalopathies (TSEs), including Kuru, variant Creutzfeldt Jakob disease (vCJD), bovine spongiform encephalopathy (BSE), chronic wasting disease (CWD), and scrapie are all examples of incurable and uniformly fatal neuro-degenerative diseases that arise from the self-propagation of misfolded prion proteins. The current working hypothesis is that small numbers of misfolded, β-rich prions (called PrPSc) are able to catalytically convert native, monomeric, helix-rich cellular prion protein (PrPC) to large numbers of misfolded prions,Citation1 which aggregate into larger, uniformly organized, oligomeric substructures that can further assemble into fibrillar, macrostructures. In this way, the misfolded prions are able to spread exponentially leading to loss of neuronal function wherever PrPC is highly expressed. PrPC is found in all mammalian cells; but the highest level of expression is in neurological tissues,Citation2,Citation3 which largely explain the neuro-degenerative nature of prion diseases.
The native prion protein, PrPC, is a lipid-bound, monomeric, ~24 kDa protein with an unstructured N-terminal region containing roughly 70 residues and a globular core consisting primarily of α-helical secondary structureCitation4 (42% α-helix and about 5% β-sheet). This unstructured N-terminus consists of 5 octa-repeat segments that have been shown to bind divalent metal ions (Cu2+, Fe2+, Zn2+).Citation5,Citation6 The core contains 2 N-linked glycosylation sites at Asn181 and Asn197Citation7,Citation8 while the C-terminus is linked to a lipid glycosylphosphatidylinositol (GPI) anchor that secures the protein to the cellular membrane. PrPSc is rich in β-sheet structure that contains 10% α-helix and 30% β-sheet.Citation1,Citation9,Citation10 However, a recent study showed that PrPSc consists largely of β-strands with no α-helices.Citation11 These β-sheets tends to form large oligomers, insoluble aggregates, and fibril like structures. As a result, PrPSc is protease-resistant, oligomeric, and substantially less soluble than PrPC. These divergent features allow the 2 isoforms to be easily differentiated biochemically.
After many decades of investigation, much is known of the prion protein's (PrPC) native structure; however, less is known about the β rich isoform. Likewise, very little is known about the α-helix to β-sheet conversion process, the precise mechanisms behind self-seeded propagation or the potential causative agents that might induce sporadic prion disease. As prion protein misfolding and aggregation is believed to be a major contributor to prion disease etiology, a more detailed molecular understanding of the ordered aggregation and amyloid fibril formation process is critical for developing strategies to prevent or treat these conditions.
To help better understand biochemical aspects of PrP conversion, several groups have used in vitro or test-tube systems in which recombinant PrPC molecules are converted into to β-rich PrPSc-like molecules (PrPβ) using a number of chemical and/or physical agents such as guanidine hydrochloride,Citation12 urea,Citation13 SDS,Citation14 high temperature,Citation15 low pHCitation16 that cause complete or partial protein denaturation. Most of these protocols not only cause the conformational change of PrPC into PrPβ but also generate “synthetic prions” that exhibit many of the physiochemical properties seen in PrPSc molecules isolated from diseased tissues.Citation17,Citation18 However, unlike brain-derived prions, the conversion and apparent propagation of these synthetic prions is dependent on continued exposure to detergents. Despite the widespread belief in the “protein-only” hypothesis concerning prion protein conversion and infectivity, controversies over whether other molecules beside PrPSc are also required for conversion and replication in vivo, still remain. In particular, the role of a number of naturally occurring, non-proteinaceous molecules such as nucleic acids,Citation19 polyanions,Citation15,Citation20 lipids,Citation21-Citation23 sulfated glycans,Citation15 and metalsCitation24 are actively being investigated in the pathogenesis of prion diseases.
Recently, it was shown that recombinant prion proteins could be rendered seed-competent (and infectious) with the addition of total liver RNA and synthetic 1-palmitoyl-2-oleoyl-phosphatidylglycerol (POPG) lipid molecules.Citation20 Even more recently it was reported that phosphatidylethanolamine (PE) is an essential cofactor for generating infectivity from converted recombinant PrP.Citation25 These findings strongly suggest that a non-protein molecular component may be responsible for facilitating or augmenting the prion protein conversion process.Citation19,Citation25 Given these emerging results, we began screening for a “prion protein converting molecule” among naturally occurring, exogenous molecules. The recent discovery that the N-terminal residues of the human prion protein (PrPC) has broad spectrum antibacterial propertiesCitation26 mediated by membrane-prion protein interactions directed our screen toward analyzing bacterial components and specifically looking at lipopolysaccharide (LPS), an outer membrane component of gram-negative bacteria. In particular, LPS is a temperature resistant, amphipathic molecule that consists of both lipid (lipid A) and carbohydrate (core and O-antigen) components. LPS acts as an immunogen and a strong pyrogen in healthy mammals. It binds the CD14/TLR4/MD2 receptor complex which resides in lipid rafts where the prion protein co-localizes.Citation27 LPS promotes the secretion of pro-inflammatory cytokines in many cell types, but especially in macrophages. Given that both PrPC and the LPS/CD14/TLR4 receptor complex are localized to the same membrane domains via their GPI anchors and given the structural similarity of LPS to polyanionic molecules like POPG that have been associated with prion protein conversion, we decided to investigate LPS's potential effects on ShPrP (90–232) in vitro.
Interestingly, we found that native, recombinant prion protein incubated with modest concentrations of LPS (15 µg/mL) at physiological normal pH and temperature led to the rapid conversion to oligomers that were PK resistant and rich in β-sheet structure (PrPβ) compared with the native ShPrP (90–232). Furthermore this conformational conversion to a β-sheet rich structure occurs at ratios of PrP to LPS below 1:1 (w/w), as long as the LPS concentration in the conversion solution remains above the critical micelle concentration (CMC). Even more interestingly, this conversion phenomenon, whereby conversion to a β-sheet rich structure occurred down to a weight ratio of ShPrP to LPS of 1:0.09 (w/w), was not seen with other known prion conversion reagents (including SDS, urea, POPG, or PE). Given the ubiquity of LPS in both recombinant prion preparations and the abundance of LPS in many animal pens, cages, and feed, these results may have some interesting implications regarding prion conversion – both in vitro and in vivo.
Results
LPS mediated conversion
The strong interaction between the prion protein and bacterial LPS was evident from our initial binding studies performed by NMR (). Apart from a few N-terminal and 6x-His affinity tag residues, immediate and complete attenuation of ShPrP (90–232) 15N-HSQC amide signals resulted upon addition of a 1:1 ratio (w/w) of LPS to the ShPrP (90–232) sample. This interaction was also visualized by transmission electron microscopy (). As seen in , the typical cylindrical bicellular structure adopted by LPS in water was disrupted leading to a spherical micelle formation () with micelle diameters ranging between 50 and 150 nanometers. The negative staining achieved with uranyl acetate suggests that protein is adhering to the micelle surface. More interestingly, conversion to a fibril state was observed upon incubation of this complex at 37 °C (). These converted prion protein fibrils ranged in length from 100 nm to 1 µm and were associated in pairs and clustered with the LPS micelles. To further characterize this state, a circular dichroism (CD) spectrum was collected () and a proteinase K (PK) digestion assay was performed (). The CD spectrum revealed a high level of β-sheet structure and the PK digestion revealed a 12 kDa proteolytic resistant core, consistent with both β-oligomericCitation28 and spontaneous PMCA-derived isoforms.Citation29 To test if this phenomenon was restricted to Syrian hamster prion protein we also found that mixing pure LPS in the presence of NaCl (150 mM) with recombinant mouse prion protein at mg ratios (w/w) of 1:1 (LPS:ShPrP (90–232) also resulted in instant conversion to PrPβ (result not shown).
Figure 1. Region of the 15N-HSQC NMR spectra for the ShPrP90–232 before (A) and after (B) the addition of an equimolar amount of LPS.
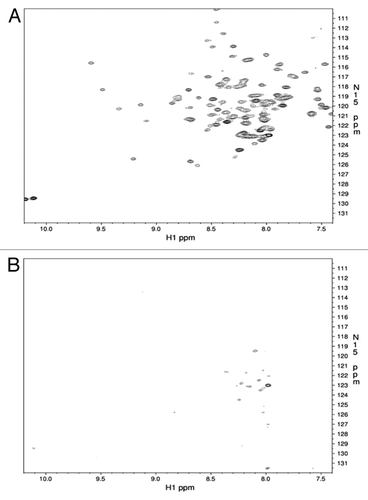
Figure 2. Electron microscope images of the LPS mediated conversion of the ShPrP (90–232) protein. (A) LPS micelles at 0.5 mg/mL in water, pH 7.0 (magnification 71 000×, scale bar 50 nm). (B) The same LPS “micelles” shown in (A) after addition of 0.5mg/mL ShPrP (magnification 56 000×, scale bar 200 nm). (C and D) The sample after the addition of 150 mM NaCl, 0.1% NaN3 and incubated at 37 °C for 48 h magnification 110 000× and 14 000×, scale bars: 50 nm and 0.5 um respectively). Inset in was collected at 110 000× (scale bar 200 nm). Small fibrils can be seen in C. Samples were stained with lead acetate (A) or uranyl acetate (B–D).
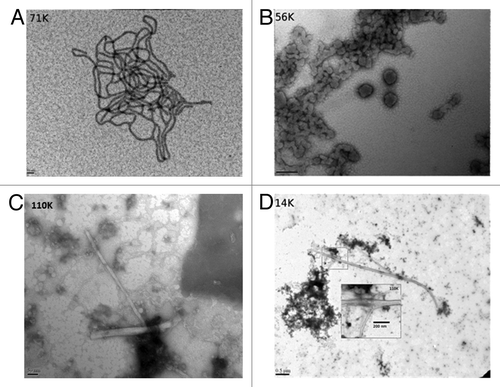
Figure 3. (A) SDS-PAGE gel of proteinase-K treated ShPrP isoform.Lane 1: LPS-generated ShPrPβ. Lane 2: ShPrP (90–232). Lane 3: ShPrP digested with PK (PK:ShPrP 1:1000). Lane 4: Molecular weight ladder (molecular weights on the right). Lane 5: PK:ShPrPβ 1:1000. Lane 6: PK:ShPrPβ 1:750. Lane 7: PK:ShPrPβ 1:500. Lane 8: PK:ShPrPβ 1:250. (B) CD spectra for the ShPrP (90–232) with 1:1 w/w ratio of LPS at 0 incubation time.
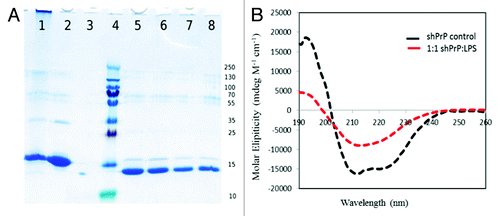
These preliminary findings warranted a more in-depth characterization of the LPS mediated conversion process. In particular we decided to investigate the effect of differing LPS concentrations on the prion conversion process. shows the results of the LPS-mediated conversion in which solutions with progressively lower ShPrP (90–232) to LPS ratios were prepared and monitored by CD. Weight (w/w) ratios above 1:0.5 (ShPrP (90–232):LPS) resulted in the complete conformational change of PrPC into β-sheet oligomers. Meanwhile, solutions in which the LPS was below the critical micelle concentration (CMC) of 14 µg/mL did not show any conversion at all, even after 4 wk of incubation. Thus we conclude that a micellar form of LPS (as might be found in a live or lysed bacterial cell) is required for initiating conversion. The secondary structure content (% α-helix and β-sheet) of the prion proteins were calculated from the CD data presented in for each dilution and are shown in .
Figure 4. CD spectra for 25 μM of ShPrP (90–232) with various concentration (w/w ratios) of LPS (1:6 300 μM LPS, 1: 3–150 μM LPS, 1:1.5–75 μM LPS, 1:0.75–37.5 μM LPS, 1:0.38–18.5 μM LPS, 1:0.17–9.5 μM LPS, 1: 0.09–5 μM LPS, and 1:0.01–1 μM LPS). Spectra were collected immediately after adding LPS, except for the 1:0.01 w/w ratio of LPS with shPrP(90–232), which was incubated an additional 4 wk to ascertain any changes in secondary structure.
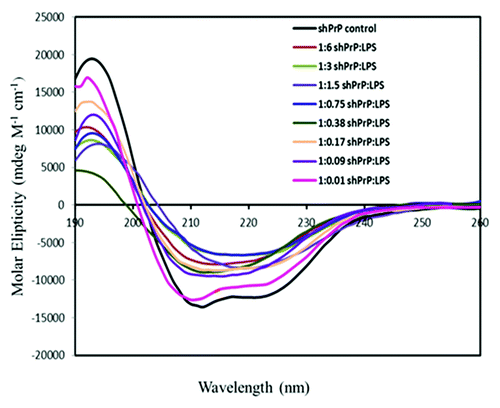
Table 1. Secondary structure content (measured by CD) of LPS-converted prions
Serial dilution
Solutions containing LPS, unconverted recombinant ShPrP (90–232), and LPS-converted PrPβ (in various concentrations) was also used to assess the unique concentration dependence property of LPS in PrP conversion. Mixing pure LPS in the presence of NaCl (150 mM) with recombinant ShPrP (90–232) at a ratio (w/w) of 1:6 (ShPrP (90–232):LPS) resulted in instantaneous conversion to a PrPβ isoform (measured by the percentage of helical and β-sheet structure determined by CD). After confirming the conversion, the LPS-containing solution (with PrPβ) was serially diluted by half, by adding equal volumes of fresh recombinant ShPrP (90–232) of the same concentration to the LPS converted PrPβ. This serial dilution process generated subsequent solutions with ShPrP (90–232) to LPS ratios (w/w) of 1:6, 1:3, 1:1.5, 1:0.75, 1:0.38, 1:0.17, 1:0.09, and 1:0.01 (; ). Interestingly conversion of ShPrP (90–232) to a β-sheet rich structure (above 20% β-sheet) was maintained even when the molar ratio (mol/mol) of LPS to ShPrP (90–232) fell below 1:1 (equivalent to 1:0.38 w/w ratio of ShPrP (90–232):LPS). This is unlike other conversion methods that only showed a dilution effect upon addition of fresh ShPrP (90–232). This confirmed that a direct interaction and conversion was being observed between LPS and the protein. It also confirmed that the measured conformational change was not simply due to a denaturation effect. This conversion was observable at all LPS concentrations, until the LPS concentration dropped below its CMC (ShPrP (90–232):LPS ratio (w/w) of 1:0.01). This effect is likely due to the sensitivity of the experimental method used to detect the change. More importantly, this serial dilution effect was used with RENAGE gel analysis (discussed below) to ascertain the molecular interaction ratio.
LPS-mediated PrPβ conversion and its subsequent serial dilution was compared with the conversion and serial dilution mediated by other chemical denaturants: namely SDS, POPG, DOPE, and urea (; ). While LPS, SDS, POPG, and PE all share lipid moieties, only PE has a positive charge or a cationic group provided by a primary amine. Furthermore, while SDS and POPG both contain negatively charged polar heads only LPS contains a phosphate group along with a complex saccharide. LPS-generated PrPβ demonstrates a capacity to convert prion protein even at sub molar ratios as the concentration of LPS is reduced (down to its CMC) in contrast to all other reagents which show a progressive dilution effect. In other words, as the concentration or urea, POPG, and SDS is reduced, the extent of conversion is reduced. Interestingly SDS can only convert ShPrP (90–232) to PrPβ at concentrations below its CMC of 8 mM (0.23% w/v). On the other hand conversion with urea and salt is time dependent with a faster conversion below the denaturation midpoint of 3.6 M.Citation30 This suggests the importance of a partially unfolded intermediate in urea induced conversion. POPG has been shown to facilitate the conversion of recombinant PrP into an infectious strain capable of propagating in mice.Citation20 POPG contains 2 alkyl carbon chains attached to a dihydroxylated phosphate group. Like LPS, it requires concentrations above its CMC (and therefore in lipid vesicles) to convert recombinant ShPrP (90–232) to PrPβ.Citation31 However, unlike LPS, the extent of POPG conversion appears to be directly proportional to the concentration of POPG used (), as the concentration of POPG is reduced, the extent of conversion is largely reduced. Quite recently it was also reported that phosphatidylethanolamine (PE or DOPE) can act as a solitary cofactor in the propagation of recombinant PrPSc using brain homogenate and standard PMCA conditions.Citation25,Citation32 To explore this further, we tested whether the same concentrations of DOPE described in the original publicationCitation25 could facilitate the conversion of ShPrP (90–232) to a β-sheet conformation without brain homogenate and sonication. Interestingly, our results showed that no conversion to PrPβ occurred, even after 7 d of incubation at 37 °C (). These findings suggest that other factors, such as temperature or additional brain homogenate cofactors are responsible for initiating the conversion process mediated by PE.
Figure 5. CD spectra for the ShPrP (90–232) with various molar ratios of DOPE, SDS, POPG, and urea at 0 incubation time. (A) CD spectra of the prion propagation studies using DOPE (diluted from 1:64 to 1:16 PrPC:DOPE (w/w)). (B) Urea induced oligomers and fibrils (for comparison of secondary structure content). (C) SDS (1:0.2 to 1:0.0125 PrPC:SDS). D. POPG (diluted from 1:0.5 to 1:0.065 of ShPrP [90–232]:POPG). Each diluted sample was incubated an additional 19 d to ascertain any changes in secondary structure.
![Figure 5. CD spectra for the ShPrP (90–232) with various molar ratios of DOPE, SDS, POPG, and urea at 0 incubation time. (A) CD spectra of the prion propagation studies using DOPE (diluted from 1:64 to 1:16 PrPC:DOPE (w/w)). (B) Urea induced oligomers and fibrils (for comparison of secondary structure content). (C) SDS (1:0.2 to 1:0.0125 PrPC:SDS). D. POPG (diluted from 1:0.5 to 1:0.065 of ShPrP [90–232]:POPG). Each diluted sample was incubated an additional 19 d to ascertain any changes in secondary structure.](/cms/asset/941c5b2a-f225-4a74-98eb-4cf229092a26/kprn_a_10928939_f0005.gif)
Table 2. Secondary structure content of urea, DOPE, POPG, and SDS mediated conversion of prion protein
Given that the previous propagation studies of both DOPE and POPG were used in conjunction with other cellular components (cell extracts, RNA), it suggests that additional agents are required to mediate the conversion of PrPC to a propagating isoform. Our findings suggest that saccharide moieties (which are found only in LPS) may have the necessary requirements to facilitate conformational conversion.
Size comparison of PrPβ formed by conversion and fibril propagation
To compare the size of PrPβ aggregates formed by the various prion protein conversion techniques we used a native gel electrophoresis technique called RENAGE.Citation33 With RENAGE it has been previously shown that the PrPβ oligomers formed via urea/salt conversion consist of a distribution of heptamers to dodecamers.Citation33 This size distribution of medium-sized oligomers is seen in . In contrast, LPS-converted prion protein (PrPβ) are characterized by very large aggregates that migrate just a few millimeters into the stacking gel. The large size of these oligomers may be due to the presence of tightly bound LPS. Quantifying the protein bands for the LPS-converted prion proteins at 1:0.09 PrP:LPS shows that 19% of PrP is found in the large aggregate band, 15% is found in oligomers, 6% is in the dimer band and the rest is in the monomer band. Given that the molecular weight of monomeric LPS is roughly half that of the prion protein, the 1:0.09 weight (w/w) ratio equates to a 1:0.18 molar (mol/mol) ratio. This correlates strongly with the 19% protein band shown in RENAGE. An equimolar component of the protein is found in the oligomers. We propose that these oligomers are loosely associated with the tightly bound protein. More importantly, this loosely bound protein, while dissociable from the micellar bound component, still persists in a β-sheet rich, oligomeric state. Based on a two-state model, the native α-helical prion protein (ShPrP (90–232) has an α helix content of 42% and β-sheet content 9% while the LPS-converted PrPβ has only 10% α helix and 32% β-sheet structure (). If only the oligomeric, LPS-bound 18% of protein was in fact converted, the resulting CD spectra of the 2 states should present CD spectra showing 36% helix and 14% β sheet. Instead, a calculated CD spectra for 40% converted PrPβ protein and 60% unconverted (ShPrP [90–232]) monomer would result in a CD spectra with 29% α helix and 18% β sheet composition, which is close to the observed values (). Thus, twice the amount of β-sheet structure in LPS-converted PrPβ than predicted for a 1:0.18 ShPrP (90–232):LPS molar ratio, indicates a stoichiometric interaction ratio of 2:1 ShPrP (90–232):LPS.
Figure 6. Resolution enhanced native acidic gel electrophoresis (RENAGE) shows different size and stability of oligomers formed from the different conversion methods. Conversion of recombinant ShPrP (90–232) is compared for urea/salt conversion (3M urea 20 mM sodium acetate pH 4 and 200 mM NaCl), POPG conversion (0.05:1 POPG to PrP [w/w], incubated at 37 °C for 4 d), SDS conversion (0.02% SDS), and LPS conversion (1:0.09 PrP to LPS, incubated at 37 °C for 2 d), 8 μg of each sample is loaded. The latter is from MoPrP (90–231) cross-linked non-specifically using PICUP.
![Figure 6. Resolution enhanced native acidic gel electrophoresis (RENAGE) shows different size and stability of oligomers formed from the different conversion methods. Conversion of recombinant ShPrP (90–232) is compared for urea/salt conversion (3M urea 20 mM sodium acetate pH 4 and 200 mM NaCl), POPG conversion (0.05:1 POPG to PrP [w/w], incubated at 37 °C for 4 d), SDS conversion (0.02% SDS), and LPS conversion (1:0.09 PrP to LPS, incubated at 37 °C for 2 d), 8 μg of each sample is loaded. The latter is from MoPrP (90–231) cross-linked non-specifically using PICUP.](/cms/asset/e7614a27-003c-4864-aa44-3c265784b992/kprn_a_10928939_f0006.gif)
RENAGE was also used to compare oligomer sizes and structure induced by POPG and SDS. As seen in , RENAGE of the POPG-converted prion protein (PrPβ) shows only a small amount of aggregation. The POPG-converted oligomers appear to be unstable under native electrophoresis. Similarly, conversion of ShPrP (90–232) with 0.02% w/v SDS resulted in large aggregates that appear to dissociate as electrophoresis proceeds. While both the SDS and POPG conversion techniques produced β-rich oligomers with about 30% β sheet content (), only the POPG formed oligomers could dissociate into a monomeric state, while the SDS formed PrPβ remained in a loosely associated oligomeric state. Efforts to generate PrPβ oligomers or fibrils using DOPE were not successful and so they were not studied by RENAGE. In contrast, the LPS generated PrPβ is distributed into various states (). We also found that mixing LPS with recombinant cervid prion protein or recombinant mouse prion protein at a weight ratio of 1:1 LPS:ShPrP (90–232) (w/w) also resulted in conversion to large aggregates (result not shown).
Nucleic acid control experiments
Discrepancies exist in the literature regarding the necessary components required to induce conversion of the prion protein into a self-replicating form.Citation15,Citation20,Citation23,Citation34,Citation35 While cofactors are not necessary for converting recPrPC to PrPSc in prion seeded PMCA,Citation36 cellular components like RNA and lipids are required to generate de novo, self-propagating prions from recombinant prion protein sources.Citation19,Citation20 This issue needed to be addressed given that the commercial sources of LPS are contaminated with various amounts of nucleic acids (RNA, DNA, short oligos, and free NTPs). To rule out the potential effects that certain bacterial cellular components from E. coli might have on our observed reactions we sought to control for these known LPS contaminants. While RNA is reported to interact with N-terminal residues of PrP,Citation35,Citation37 DNA has been shown to interact with the C-terminal residues of the prion protein.Citation34 Given that our recombinant prion protein construct is devoid of any reported RNA binding site and that our lab does not control for RNase activity, the influence that RNA might have on the observed conversion was of less concern than that of DNA. Two sources of LPS were obtained from Sigma. One preparation (L3012) is purified by gel filtration and is reported to have >10% nucleic acid contamination, while the other (L3024) is further purified by ion-exchange chromatography and contains <1% nucleic acid. The prion protein conversion reactions were repeated with the 2 different LPS preparations with no significant difference in the conversion rate observed (data not shown). Furthermore, the addition of DNase and RNase to the LPS stock solution (L3024) prior to initiating the reaction also yielded similar conversion rates. These data strongly suggest that nucleic acid contamination is not the cause of the observed LPS-mediated conversion.
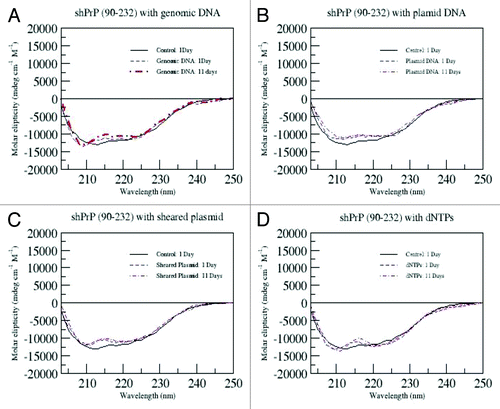
However, given that residual nucleic acid fragments would still be present in the reaction, additional experiments with various forms of DNA (dNTP's, sheared plasmid, whole plasmid, and genomic E. coli DNA) added to the conversion solution were also performed. These CD data are shown in . These controls were followed for 11 d. Apart from the expected CD spectral changes due to the addition of nucleic acids, no significant change in secondary structure of the protein was observed throughout the course of the experiment. Thus, we conclude that the observed changes in converted prion protein (PrPβ) secondary structure arose from LPS alone and that LPS, alone, is responsible for initiating the PrP conversion reactions and that nucleic acids do not mediate the conversion or influence the LPS-directed conversion.
Figure 7. CD spectra for ShPrP (90–232) mixed with 1:1 molar ratios of with either genomic DNA, plasmid DNA, sheared plasmid DNA or dNTP's in all experiments. The samples were incubated for the indicated time.
Discussion
In this study we have shown that LPS interacts with recombinant prion protein and that this interaction converts the α-helix-rich recombinant ShPrP (90–232) into a β-rich, PK-resistant form (PrPβ) under physiological conditions. LPS induced PrPβ conversion does not require any brain-derived PrPβ seed, additional co-factors, or denaturants. This is in marked contrast to most other conversion and propagation protocols.Citation14,Citation16,Citation20,Citation29,Citation38,Citation39 Furthermore, many published conformational prion protein conversion strategies rely on a molar excess or destabilizing amounts of chemical denaturants (urea, guanidine hydrochloride), synthetic detergents (SDS), or extremes of pH or high (denaturing) temperatures.Citation12-Citation16 However, LPS-mediated prion protein conversion is distinct from these methods in that measurable conversion to a β-sheet rich form occurs at ShPrP (90–232) to LPS ratios down to 1:0.09 (w/w). Additionally, sizing of oligomers using RENAGE shows that the LPS converted PrPβ oligomers differ from those generated by POPG, SDS, and urea. Furthermore, RENAGE data showed that the LPS-generated PrPβ oligomers will dissociate to monomers. This differs from PrPβ oligomers formed by urea () or acid,Citation33 which are stable under electrophoresis conditions.
These findings may have significant consequences for laboratories using recombinant prion proteins in PMCA or in vitro conversion assays and for laboratories involved with animal prion transmission experiments. In particular, an excess of residual LPS may skew conversion of recombinant PrP to a positive result. Furthermore, most research laboratories, dissection rooms, and animal housing facilities are heavily contaminated with LPS.Citation40-Citation42 Indeed, unless a facility is maintained under the strictest decontamination protocols, LPS contamination is almost certain. While we have demonstrated the converting abilities of LPS in vitro, these findings do not necessarily translate into propagating (or infectious) prions in animal models. Animal studies with LPS and LPS-converted prion protein are currently ongoing and are yielding interesting preliminary results. However, these will be the subject of another manuscript. As discussed earlier, our findings indicate LPS concentrations above the CMC is required to detect prion protein conversion. How and when such a micellar form exists in nature and whether induces sporadic prion disease is certainly worth some further investigation. These findings partially support the hypothesis that prion-associated diseases may be the result of, or augmented by, the presence of LPS-containing bacteria.
LPS is present in the saliva, mammary glands, vagina and postpartum uterus, gastrointestinal tract, and feces of most mammals, including sheep, elk, deer, and cattle. LPS is a key component of the normal Gram-negative bacterial flora of all mammals. In the case of cattle, it has been documented that the levels of LPS in the gastrointestinal tract (i.e., the rumen) increases significantly when they are put on high grain diets compared with when they are left on pastures.Citation43-Citation45 One studyCitation46 followed cattle fed on grain pellets comprised of 50% wheat and 50% barley. After 4 d on this diet, free ruminal LPS measured 12.8 × 104 EU/mL or 12.8 µg/mL, which is very close to LPS's CMC. Similar LPS values would likely be found in other ruminants (elk, deer, sheep) consuming similar high grain diets. It should also be noted that these values reflect free LPS, not bound material in living bacteria. In terms of bound LPS, it has been estimated that each E. coli bacterium contains between 1 and 2 million individual LPS molecules, which comprise over 70% of the its outer membrane. This corresponds to a LPS concentration of between 120 and 240 µg/mL. If normal prion protein were to come into contact with these high LPS concentrations in the gut (through consumption of meat or bone meal or eating prion-rich tissues), it is possible that the prion protein could convert to β oligomeric forms. LPS is not restricted to the gut. It can also be found in blood and tissues of mammals too, especially those suffering bacterial infections such as septicemia or meningitis.Citation47 Another possible route for prion protein-LPS association could be through contact with dead, dissected, or slaughtered animals, which are decaying or awaiting rendering. Bacteria proliferate quite rapidly in such circumstances. Given that LPS is resistant to heat treatment,Citation48 both chemical (1M NaOH) and thermal degradation is required for its complete removal. Interestingly, prior to the BSE outbreak in the UK (ca. 1987) and the ban on meat and bone meal in animal feed, carcasses were wet rendered with only minimal heating (95 °C).Citation49-Citation51 Consequently one could conclude that high levels of LPS must have been present in meat and bone meal (MBM) in the 1980s and 1990s. However, despite these relatively high levels of LPS in everyday occurrence, we do not see high levels of sporadic prion disease. This suggests that while LPS may interact strongly with ShPrP (90–232) and initiate conversion, the LPS-converted PrPβ may not be particularly toxic. Additionally, because the PrP-LPS interaction appears to exhibit specificity at stoichiometric ratios and induces a conformational change in the protein, it likely has biological significance, which may or may not influence the pathology of prion associated diseases.
In conclusion, we have shown that LPS, a naturally occurring, ubiquitous agent found in almost all animals, is able to convert recombinant prion protein into PrPβ oligomers under normal, physiological conditions. The β-rich form of the prion protein that is produced by LPS is morphologically similar to that produced by another anionic lipid, POPG. However, the specific component of LPS (endotoxin, lipid A, or polysaccharide) that actually interacts with the protein or initiates conversion remains to be determined. Studies to determine this and whether these synthetic prions are propagation-competent and/or neurotoxic in animal models are ongoing.
Materials and Methods
Materials
For the experiments described here, several recombinant mammalian prion protein variants were studied, including Syrian hamster, mouse, and cervid prion protein. All of the studies were conducted with the recombinant Syrian hamster prion protein initially and later the results were confirmed using recombinant mouse and cervid prion protein. Since the methods and results were essentially identical for all mammalian prion protein studied, to reduce space and prevent duplication, we shall focus on describing the methods and results from the Syrian hamster prion protein only. The expression and purification of a truncated form (residues 90–232) of the Syrian hamster prion protein (ShPrP) linked to a 22-residue N-terminal fusion tag containing a 6x-His and a thrombin cleavage site has been described elsewhere.Citation16 LPS from Escherichia coli O111:B4 (L3012: 10–20% nucleic acid; L3024: <1% nucleic acids), SDS was purchased from Sigma-Aldrich (Cat# L3771) and proteinase K was from Promega (V3021). Palmitoyl-oleoyl-phosphatidyl-glycerol or POPG (catalog number 840457) and Dioleoyl-phosphatidyl-ethanolamine or DOPE (catalog number 852758P) were acquired from Avanti Polar Lipids.
LPS, SDS, POPG, and urea induced conversion of recombinant PrP to PrPβ
Lyophilized LPS was reconstituted with milliQ H2O to provide an initial working concentration of 5 mg/mL. To assess LPS-mediated prion protein conversion and concentration dependence, this stock solution was diluted and used to reconstitute lyophilized ShPrP (90–232) to ~0.5 mg/mL (500 μL in 1.5 mL eppendorf tubes), providing ratios of shPrP:LPS (w:w) of 1:6, 1:3, 1:1.5, 1:0.75, 1:0.38, 1:0.17, 1:0.09, and 1:0.01. Sodium chloride (150 mM) and 0.1% NaN3 were subsequently added to each sample. The samples were incubated (without shaking) at 37 °C for 1 wk. Given that the average molecular weight of LPS is about 10 kDa or half that of the prion protein, these weight ratios equate to twice that of the molar ratio. Because an exact molecular weight does not exist for LPS, weight ratios were used in this study for comparison and consistency.
SDS, urea, POPG, and DOPE directed prion protein conversions were done using the same protocol outlined for LPS. For SDS, a 10% stock solution was prepared and diluted into a prion protein solution (0.5 mg/mL or 26 μM) leading to a final SDS concentration of 0.02% w/v (1:0.4 ShPrP [90–232]:SDS w/w ratio; ~1:27 mol/mol ratio). Subsequently, for propagation experiments, this converted material was diluted with fresh shPrPC(90–232) such that the final SDS w/v concentrations were 0.01%, 0.005%, 0.0025%, 0.00125%, and 0.000625%. These values correspond to ratios of ShPrP (90–232):SDS (w/w) of 1:0.2, 1:0.1, 1:0.05, 1:0.025, and 1:0.0125.
For POPG mediated conversion and concentration dependence experiments, a stock suspension of 1 mg/mL (1.3 mM) in 20 mM NaHPO4 (pH 7.2, 150 mM NaCl with 0.01% NaN3) was prepared and added to the protein (0.4 mg/mL) providing ShPrP (90–232):POPG ratios (w/w) of 1:0.5 (1:12 mol/mol ratio). Subsequent dilution of the converted PrP with fresh ShPrP (90–232) provided the weight ratios of 1:0.25, 1:0.125, and 1:0.0625.
For DOPE mediated conversion experiments, a stock suspension of 20 mg/mL in 0.04% Triton-X100 (sonicated for 30 min) was prepared and added to the protein (0.4 mg/mL) providing ShPrP (90–232): DOPE ratios (w/w) of 1:65 (~1:1700 molar ratio), 1:32.5, and 1:16.25 in the presence of 150 mM NaCl with 0.01% NaN3 . The experiments were performed in 1.5 mL Eppendorf tubes with 0.5 mL of sample volume.
Urea-induced oligomerization of the prion protein was performed by reconstituting lyophilized ShPrP (90–232) prion protein into 20 mM sodium acetate buffer containing 3 M urea and 200 mM NaCl at pH 4.0. To form fibrils under denaturing conditions, a 100 µM stock solution of previously denatured ShPrP (90–232) in 6 M guanidinium chloride was diluted to 20 µM in 50 mM Hepes buffer (1 M guanidinium chloride, 3 M urea, 150 mM NaCl, pH 7.0) and shaken at 500 RPM for 18 h prior to dialyzing into 10 mM sodium acetate pH 5.2.
CD spectroscopy and data processing
The prion protein conversion reactions were followed using CD spectroscopy. CD spectra were collected on the samples within an hour of LPS addition and 1 wk later following incubation at 37 °C. Spectra were recorded in the far-UV region (190–260 nm) at 25 °C in a 0.02 cm path-length quartz cell on an Olis DSM 17 spectropolarimeter. Five scans were averaged for each CD spectrum collected. The protein concentrations were 0.5 mg/mL (~25 µM). Blank spectra were collected in the same manner and subtracted from the measured prion protein spectra prior to molar ellipticity determination. Ellipticity values were calculated using an average amino acid molecular weight of 113.64 g/mol and the secondary structure content was calculated with CDProCitation52 using the CONTINLL algorithm and the SP22X reference set.Citation53
Transmission electron microscopy
Typically 4 µL of each prion protein or prion protein-LPS sample were spotted onto 300 mesh copper EM grids (TED PELLA INC., Cat# 01813-F) and allowed to adsorb for 5 min. The remaining sample was wicked away with a kimwipe and the grid air-dried for 5 min, rinsed with 5 μL of milliQ H2O, and then negatively stained for 1 min with 4 μL of either 1% lead acetate for LPS-containing samples or 4% uranyl acetate for samples containing the prion protein. The excess staining solution was wicked away and the samples were re-rinsed with another 5 μL of mqH2O and dried for 5 min. The grids were analyzed on a Philips/FEI (Morgagni) transmission electron microscope (TEM) to look for and assess the structure and morphology of PrP oligomers and fibrils.
Proteinase-K (PK) resistance assay
The method used to assess proteinase-K (PK) resistance of the LPS-converted or template converted prion protein was adapted from Bessen and Marsh.Citation54 Specifically, proteinase K (Promega) was dissolved in 50 mM TRIS-HCl (pH 8.0), 2 mM CaCl2 to a concentration of 2.5 mg/mL. Urea was added to each protein sample to a final concentration of 3 M, and the samples were then incubated for 30 min at 37 °C in the presence of various proteinase K concentrations (250:1, 500:1, 750:1, and 1000:1 prion:PK ratios). Protein digestion was stopped with the addition of 5 mM phenylmethanesulfonyl fluoride (PMSF) and the reactions analyzed using 12% SDS PAGE stained with Coomassie blue.
Nucleic acid control experiments
Because LPS contains some level of nucleic acid contamination and because it was unclear whether nucleic acids were contributing to the observed prion protein conversion, we performed additional control experiments with genomic DNA, DNA fragments, and dNTPs. Genomic DNA was purified from E. coli (BL21DE3) using a Maxiprep kit (Qiagen, Cat# 12163) while dNTP's were acquired from Fermertas (Thermo Scientific, Cat# R0192) and mixed to equimolar ratios prior to use. The source of plasmid DNA was the pET15b+ vector constructed with the ShPrP (90–232) gene used for expression in this study. This plasmid was also fragmented mechanically by raking 10 µL of the plasmid in a 1.5 mL Eppendorf tube over a 90 well Eppendorf flotation rack to provide a source of sheared DNA. Visual confirmation of the random fragmentation was obtained by running the DNA sample on a standard 2% agarose gel prior to use. The concentration of the polynucleic acid was quantified on a Nanodrop ND-1000 spectrophotometer. The molar ratios of protein to nucleic acids were 1:1 in all experiments. The samples (500 μL with 0.1% NaN3) were incubated at 37 °C in 1.5 mL Eppendorf tubes and analyzed by CD spectroscopy at 24-h intervals for 11 d. For additional control experiments, residual nucleic acids in the LPS samples (L3024) were also pretreated with DNase and RNase. The LPS stock solution (5 mg/mL in mqH2O) was diluted to 1 mg/mL with the addition of 20 mM TRIS-HCl (pH 8.3). MgSO4 and CaCl2 were added to concentrations of 2 mM for the RNase and DNase samples, respectively, prior to adding the enzymes at working concentrations of 0.2 µg/mL. The resulting stock solutions were left to react at 37 °C for 18 h prior to use.
NMR analysis
Recombinant ShPrP (90–232) was expressed in minimal media with 15N ammonium chloride and purified as previously described.Citation16 The sample was dialyzed into 20 mM sodium acetate buffer (pH 7.0) with 150 mM NaCl and 0.1% NaN3 and concentrated using a 3000 MW cutoff Centricon device (Millipore Inc., Cat# UFC903024). Ten percent D2O was added for solvent lock and the sample was transferred to a Shigemi NMR tube. The final concentration was 620 µM (320 µL with 30 µL of D2O). 15N-HSQC spectra were collected on a 500 MHz Varian INOVA NMR fitted with a z-gradient, triple resonance probe. A total of 1024 points were collected in the t2 dimension with a sweep width of 6000 Hz and 96 points were collected in the t1 dimension with a spectral width of 1800 Hz. Immediately following collection of the control experiment, LPS (123 µL of a 5 mg/mL aqueous solution) was added to achieve roughly a 1:1 molar ratio. The final protein concentration was 460 µM. This spectrum was recollected with the same parameters outlined above. Forty transients were collected and averaged for each spectrum.
RENAGE gel electrophoresis
Separation of prion protein oligomers by RENAGE was performed as described previously.Citation33 Briefly, converted prion protein (8 µg) was loaded onto an 8% RENAGE gel at pH 4.3 with a 3% stacking gel at pH 5.2. The gels were pre-run for 20 min prior to loading protein samples. Separation was obtained with a constant current of 30 mA for 72 min. The gel was stained with colloidal Coomassie blue for 4 h and de-stained with water.Citation55 Gel lanes were converted to a band-intensity chromatogram using ImageJ (http://rsbweb.nih.gov/ij/index.html). Chromatograms were then converted to text file and plotted in Origin software. The peaks were then manually chosen and integrated, assuming Gaussian peaks. Percentages were then calculated from the area of the peak compared with the total integrated area.
| Abbreviations: | ||
| TSE | = | transmissible spongiform encephalopathy |
| CJD | = | Creutzfeldt Jakob disease |
| BSE | = | bovine spongiform encephalopathy |
| CWD | = | chronic wasting disease |
| PMCA | = | Protein Misfolding Cyclic Amplification |
| EM | = | electron microscopy |
| TEM | = | transmission electron microscopy |
| PK | = | proteinase K |
| PrP | = | prion protein |
| ShPrP | = | the core alpha helical Syrian hamster prion protein residues 90-232 |
| ShPrPβ | = | beta-sheet converted Syrian hamster prion protein (90-232) |
| PrPC | = | cellular prion protein |
| LPS | = | lipopolysaccharide |
| POPG | = | 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-(1'-rac-glycerol) |
| SDS | = | sodium dodecyl sulfate |
| PE | = | phosphatidylethanolamine |
| DOPE | = | 1,2-Dioleoyl-sn-Glycero-3-Phosphoethanolamine |
| PMSF | = | phenylmethanesulfonyl fluoride |
| CMC | = | critical micelle concentration |
| CD | = | circular dichroism |
| NMR | = | nuclear magnetic resonance |
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
This work was supported by an Alberta Meat and Livestock Agency (ALMA) grant to B.N.A. and D.S.W. and by Alberta Prion Research Institute (APRI) and PrioNet Canada grants to D.S.W. We thank Ashenafi Abera and Bow Suriyamongkol for providing purified PrP. CD spectra were acquired with the help of Wayne Moffat in the Analytical Instrumentation Laboratory. EM data was acquired with the help of Arlene Oatway in the Biological Sciences Microscopy Unit.
Related Research Data
References
- Pan KM, Baldwin M, Nguyen J, Gasset M, Serban A, Groth D, Mehlhorn I, Huang Z, Fletterick RJ, Cohen FE, et al. Conversion of alpha-helices into beta-sheets features in the formation of the scrapie prion proteins. Proc Natl Acad Sci U S A 1993; 90:10962 - 6; http://dx.doi.org/10.1073/pnas.90.23.10962; PMID: 7902575
- Kretzschmar HA, Prusiner SB, Stowring LE, DeArmond SJ. Scrapie prion proteins are synthesized in neurons. Am J Pathol 1986; 122:1 - 5; PMID: 3079955
- Moser M, Colello RJ, Pott U, Oesch B. Developmental expression of the prion protein gene in glial cells. Neuron 1995; 14:509 - 17; http://dx.doi.org/10.1016/0896-6273(95)90307-0; PMID: 7695897
- Riek R, Hornemann S, Wider G, Glockshuber R, Wüthrich K. NMR characterization of the full-length recombinant murine prion protein, mPrP(23-231). FEBS Lett 1997; 413:282 - 8; http://dx.doi.org/10.1016/S0014-5793(97)00920-4; PMID: 9280298
- Brown DR, Qin K, Herms JW, Madlung A, Manson J, Strome R, Fraser PE, Kruck T, von Bohlen A, Schulz-Schaeffer W, et al. The cellular prion protein binds copper in vivo. Nature 1997; 390:684 - 7; http://dx.doi.org/10.1038/37733; PMID: 9414160
- Walter ED, Stevens DJ, Visconte MP, Millhauser GL. The prion protein is a combined zinc and copper binding protein: Zn2+ alters the distribution of Cu2+ coordination modes. J Am Chem Soc 2007; 129:15440 - 1; http://dx.doi.org/10.1021/ja077146j; PMID: 18034490
- Rudd PM, Endo T, Colominas C, Groth D, Wheeler SF, Harvey DJ, Wormald MR, Serban H, Prusiner SB, Kobata A, et al. Glycosylation differences between the normal and pathogenic prion protein isoforms. Proc Natl Acad Sci U S A 1999; 96:13044 - 9; http://dx.doi.org/10.1073/pnas.96.23.13044; PMID: 10557270
- Stimson E, Hope J, Chong A, Burlingame AL. Site-specific characterization of the N-linked glycans of murine prion protein by high-performance liquid chromatography/electrospray mass spectrometry and exoglycosidase digestions. Biochemistry 1999; 38:4885 - 95; http://dx.doi.org/10.1021/bi982330q; PMID: 10200178
- Caughey BW, Dong A, Bhat KS, Ernst D, Hayes SF, Caughey WS. Secondary structure analysis of the scrapie-associated protein PrP 27-30 in water by infrared spectroscopy. Biochemistry 1991; 30:7672 - 80; http://dx.doi.org/10.1021/bi00245a003; PMID: 1678278
- Safar J, Roller PP, Gajdusek DC, Gibbs CJ Jr.. Conformational transitions, dissociation, and unfolding of scrapie amyloid (prion) protein. J Biol Chem 1993; 268:20276 - 84; PMID: 8104185
- Smirnovas V, Baron GS, Offerdahl DK, Raymond GJ, Caughey B, Surewicz WK. Structural organization of brain-derived mammalian prions examined by hydrogen-deuterium exchange. Nat Struct Mol Biol 2011; 18:504 - 6; http://dx.doi.org/10.1038/nsmb.2035; PMID: 21441913
- Kocisko DA, Priola SA, Raymond GJ, Chesebro B, Lansbury PT Jr., Caughey B. Species specificity in the cell-free conversion of prion protein to protease-resistant forms: a model for the scrapie species barrier. Proc Natl Acad Sci U S A 1995; 92:3923 - 7; http://dx.doi.org/10.1073/pnas.92.9.3923; PMID: 7732006
- Julien O, Chatterjee S, Thiessen A, Graether SP, Sykes BD. Differential stability of the bovine prion protein upon urea unfolding. Protein Sci 2009; 18:2172 - 82; http://dx.doi.org/10.1002/pro.231; PMID: 19693935
- Stöhr J, Weinmann N, Wille H, Kaimann T, Nagel-Steger L, Birkmann E, Panza G, Prusiner SB, Eigen M, Riesner D. Mechanisms of prion protein assembly into amyloid. Proc Natl Acad Sci U S A 2008; 105:2409 - 14; http://dx.doi.org/10.1073/pnas.0712036105; PMID: 18268326
- Wong C, Xiong LW, Horiuchi M, Raymond L, Wehrly K, Chesebro B, Caughey B. Sulfated glycans and elevated temperature stimulate PrP(Sc)-dependent cell-free formation of protease-resistant prion protein. EMBO J 2001; 20:377 - 86; http://dx.doi.org/10.1093/emboj/20.3.377; PMID: 11157745
- Bjorndahl TC, Zhou GP, Liu X, Perez-Pineiro R, Semenchenko V, Saleem F, Acharya S, Bujold A, Sobsey CA, Wishart DS. Detailed biophysical characterization of the acid-induced PrP(c) to PrP(β) conversion process. Biochemistry 2011; 50:1162 - 73; http://dx.doi.org/10.1021/bi101435c; PMID: 21189021
- Legname G, Baskakov IV, Nguyen HO, Riesner D, Cohen FE, DeArmond SJ, Prusiner SB. Synthetic mammalian prions. Science 2004; 305:673 - 6; http://dx.doi.org/10.1126/science.1100195; PMID: 15286374
- Legname G, Nguyen HO, Baskakov IV, Cohen FE, Dearmond SJ, Prusiner SB. Strain-specified characteristics of mouse synthetic prions. Proc Natl Acad Sci U S A 2005; 102:2168 - 73; http://dx.doi.org/10.1073/pnas.0409079102; PMID: 15671162
- Deleault NR, Lucassen RW, Supattapone S. RNA molecules stimulate prion protein conversion. Nature 2003; 425:717 - 20; http://dx.doi.org/10.1038/nature01979; PMID: 14562104
- Wang F, Wang X, Yuan CG, Ma J. Generating a prion with bacterially expressed recombinant prion protein. Science 2010; 327:1132 - 5; http://dx.doi.org/10.1126/science.1183748; PMID: 20110469
- Lührs T, Zahn R, Wüthrich K. Amyloid formation by recombinant full-length prion proteins in phospholipid bicelle solutions. J Mol Biol 2006; 357:833 - 41; http://dx.doi.org/10.1016/j.jmb.2006.01.016; PMID: 16466741
- Wang F, Yin S, Wang X, Zha L, Sy MS, Ma J. Role of the highly conserved middle region of prion protein (PrP) in PrP-lipid interaction. Biochemistry 2010; 49:8169 - 76; http://dx.doi.org/10.1021/bi101146v; PMID: 20718504
- Wang F, Yang F, Hu Y, Wang X, Wang X, Jin C, Ma J. Lipid interaction converts prion protein to a PrPSc-like proteinase K-resistant conformation under physiological conditions. Biochemistry 2007; 46:7045 - 53; http://dx.doi.org/10.1021/bi700299h; PMID: 17503780
- Singh N, Das D, Singh A, Mohan ML. Prion protein and metal interaction: physiological and pathological implications. Curr Issues Mol Biol 2010; 12:99 - 107; PMID: 19767653
- Deleault NR, Piro JR, Walsh DJ, Wang F, Ma J, Geoghegan JC, Supattapone S. Isolation of phosphatidylethanolamine as a solitary cofactor for prion formation in the absence of nucleic acids. Proc Natl Acad Sci U S A 2012; 109:8546 - 51; http://dx.doi.org/10.1073/pnas.1204498109; PMID: 22586108
- Pasupuleti M, Roupe M, Rydengård V, Surewicz K, Surewicz WK, Chalupka A, Malmsten M, Sörensen OE, Schmidtchen A. Antimicrobial activity of human prion protein is mediated by its N-terminal region. PLoS One 2009; 4:e7358; http://dx.doi.org/10.1371/journal.pone.0007358; PMID: 19809501
- Beutler B. TLR4 as the mammalian endotoxin sensor. Curr Top Microbiol Immunol 2002; 270:109 - 20; http://dx.doi.org/10.1007/978-3-642-59430-4_7; PMID: 12467247
- Baskakov IV, Legname G, Baldwin MA, Prusiner SB, Cohen FE. Pathway complexity of prion protein assembly into amyloid. J Biol Chem 2002; 277:21140 - 8; http://dx.doi.org/10.1074/jbc.M111402200; PMID: 11912192
- Atarashi R, Moore RA, Sim VL, Hughson AG, Dorward DW, Onwubiko HA, Priola SA, Caughey B. Ultrasensitive detection of scrapie prion protein using seeded conversion of recombinant prion protein. Nat Methods 2007; 4:645 - 50; http://dx.doi.org/10.1038/nmeth1066; PMID: 17643109
- Morillas M, Vanik DL, Surewicz WK. On the mechanism of alpha-helix to beta-sheet transition in the recombinant prion protein. Biochemistry 2001; 40:6982 - 7; http://dx.doi.org/10.1021/bi010232q; PMID: 11389614
- Kazlauskaite J, Sanghera N, Sylvester I, Vénien-Bryan C, Pinheiro TJT. Structural changes of the prion protein in lipid membranes leading to aggregation and fibrillization. Biochemistry 2003; 42:3295 - 304; http://dx.doi.org/10.1021/bi026872q; PMID: 12641461
- Deleault NR, Walsh DJ, Piro JR, Wang F, Wang X, Ma J, Rees JR, Supattapone S. Cofactor molecules maintain infectious conformation and restrict strain properties in purified prions. Proc Natl Acad Sci U S A 2012; 109:E1938 - 46; http://dx.doi.org/10.1073/pnas.1206999109; PMID: 22711839
- Ladner CL, Wishart DS. Resolution-enhanced native acidic gel electrophoresis: a method for resolving, sizing, and quantifying prion protein oligomers. Anal Biochem 2012; 426:54 - 62; http://dx.doi.org/10.1016/j.ab.2012.04.005; PMID: 22490465
- Nandi PK, Leclerc E, Nicole JC, Takahashi M. DNA-induced partial unfolding of prion protein leads to its polymerisation to amyloid. J Mol Biol 2002; 322:153 - 61; http://dx.doi.org/10.1016/S0022-2836(02)00750-7; PMID: 12215421
- Weiss S, Proske D, Neumann M, Groschup MH, Kretzschmar HA, Famulok M, Winnacker EL. RNA aptamers specifically interact with the prion protein PrP. J Virol 1997; 71:8790 - 7; PMID: 9343239
- Kim JI, Cali I, Surewicz K, Kong Q, Raymond GJ, Atarashi R, Race B, Qing L, Gambetti P, Caughey B, et al. Mammalian prions generated from bacterially expressed prion protein in the absence of any mammalian cofactors. J Biol Chem 2010; 285:14083 - 7; http://dx.doi.org/10.1074/jbc.C110.113464; PMID: 20304915
- Gomes MPB, Millen TA, Ferreira PS, e Silva NL, Vieira TCRG, Almeida MS, Silva JL, Cordeiro Y. Prion protein complexed to N2a cellular RNAs through its N-terminal domain forms aggregates and is toxic to murine neuroblastoma cells. J Biol Chem 2008; 283:19616 - 25; http://dx.doi.org/10.1074/jbc.M802102200; PMID: 18456654
- Jain S, Udgaonkar JB. Salt-induced modulation of the pathway of amyloid fibril formation by the mouse prion protein. Biochemistry 2010; 49:7615 - 24; http://dx.doi.org/10.1021/bi100745j; PMID: 20712298
- Saborio GP, Permanne B, Soto C. Sensitive detection of pathological prion protein by cyclic amplification of protein misfolding. Nature 2001; 411:810 - 3; http://dx.doi.org/10.1038/35081095; PMID: 11459061
- Gorbet MB, Sefton MV. Endotoxin: the uninvited guest. Biomaterials 2005; 26:6811 - 7; http://dx.doi.org/10.1016/j.biomaterials.2005.04.063; PMID: 16019062
- Haslett C, Guthrie LA, Kopaniak MM, Johnston RB Jr., Henson PM. Modulation of multiple neutrophil functions by preparative methods or trace concentrations of bacterial lipopolysaccharide. Am J Pathol 1985; 119:101 - 10; PMID: 2984939
- Hirayama C, Sakata M. Chromatographic removal of endotoxin from protein solutions by polymer particles. J Chromatogr B Analyt Technol Biomed Life Sci 2002; 781:419 - 32; http://dx.doi.org/10.1016/S1570-0232(02)00430-0; PMID: 12450672
- Ametaj BN, Zebeli Q, Saleem F, Psychogios N, Lewis MJ, Dunn SM, Xia JG, Wishart DS. Metabolomics reveals unhealthy alterations in rumen metabolism with increased proportion of cereal grain in the diet of dairy cows. Metabolomics 2010; 6:583 - 94; http://dx.doi.org/10.1007/s11306-010-0227-6
- Emmanuel DGV, Dunn SM, Ametaj BN. Feeding high proportions of barley grain stimulates an inflammatory response in dairy cows. J Dairy Sci 2008; 91:606 - 14; http://dx.doi.org/10.3168/jds.2007-0256; PMID: 18218747
- Gozho GN, Krause DO, Plaizier JC. Rumen lipopolysaccharide and inflammation during grain adaptation and subacute ruminal acidosis in steers. J Dairy Sci 2006; 89:4404 - 13; http://dx.doi.org/10.3168/jds.S0022-0302(06)72487-0; PMID: 17033028
- Gozho GN, Krause DO, Plaizier JC. Ruminal lipopolysaccharide concentration and inflammatory response during grain-induced subacute ruminal acidosis in dairy cows. J Dairy Sci 2007; 90:856 - 66; http://dx.doi.org/10.3168/jds.S0022-0302(07)71569-2; PMID: 17235162
- Banks WA, Robinson SM. Minimal penetration of lipopolysaccharide across the murine blood-brain barrier. Brain Behav Immun 2010; 24:102 - 9; http://dx.doi.org/10.1016/j.bbi.2009.09.001; PMID: 19735725
- Magalhães PO, Lopes AM, Mazzola PG, Rangel-Yagui C, Penna TCV, Pessoa A Jr.. Methods of endotoxin removal from biological preparations: a review. J Pharm Pharm Sci 2007; 10:388 - 404; PMID: 17727802
- Smith PG, Bradley R. Bovine spongiform encephalopathy (BSE) and its epidemiology. Br Med Bull 2003; 66:185 - 98; http://dx.doi.org/10.1093/bmb/66.1.185; PMID: 14522859
- Taylor DM. Inactivation of transmissible degenerative encephalopathy agents: A review. Vet J 2000; 159:10 - 7; http://dx.doi.org/10.1053/tvjl.1999.0406; PMID: 10640408
- Taylor DM, Woodgate SL. Rendering practices and inactivation of transmissible spongiform encephalopathy agents. Rev Sci Tech 2003; 22:297 - 310; PMID: 12793787
- Johnson WC. Analyzing protein circular dichroism spectra for accurate secondary structures. Proteins 1999; 35:307 - 12; http://dx.doi.org/10.1002/(SICI)1097-0134(19990515)35:3<307::AID-PROT4>3.0.CO;2-3; PMID: 10328265
- Sreerama N, Woody RW. On the analysis of membrane protein circular dichroism spectra. Protein Sci 2004; 13:100 - 12; http://dx.doi.org/10.1110/ps.03258404; PMID: 14691226
- Bessen RA, Marsh RF. Distinct PrP properties suggest the molecular basis of strain variation in transmissible mink encephalopathy. J Virol 1994; 68:7859 - 68; PMID: 7966576
- Neuhoff V, Stamm R, Eibl H. Clear background and highly sensitive protein staining with coomassie blue dyes in polyacrylamide gels - a systematic analysis. Electrophoresis 1985; 6:427 - 48; http://dx.doi.org/10.1002/elps.1150060905