
Abstract
It is now 18 years since the first identification of a case of vCJD in the UK. Since that time, there has been much speculation over how vCJD might impact human health. To date there have been 177 case reports in the UK and a further 51 cases worldwide in 11 different countries. Since establishing that BSE and vCJD are of the same strain of agent, we have also shown that there is broad similarity between UK and non-UK vCJD cases on first passage to mice. Transgenic mouse studies have indicated that all codon 129 genotypes are susceptible to vCJD and that genotype may influence whether disease appears in a clinical or asymptomatic form, supported by the appearance of the first case of potential asymptomatic vCJD infection in a PRNP 129MV patient. Following evidence of blood transfusion as a route of transmission, we have ascertained that all blood components and leucoreduced blood in a sheep model of vCJD have the ability to transmit disease. Importantly, we recently established that a PRNP 129MV patient blood recipient with an asymptomatic infection and limited PrPSc deposition in the spleen could readily transmit disease into mice, demonstrating the potential for peripheral infection in the absence of clinical disease. This, along with the recent appendix survey which identified 16 positive appendices in a study of 32 441 cases, underlines the importance of continued CJD surveillance and maintaining control measures already in place to protect human health.
Abbreviations
| BSE | = | bovine spongiform encephalopathy |
| CWD | = | chronic wasting disease |
| GSS | = | Gerstman–Sträussler–Scheinkerdisease |
| M | = | methionine |
| PPS | = | pentosan polysulphate |
| PrPres | = | protease-resistant prion protein |
| PrPSc | = | abnormal prion protein |
| QuIC | = | quaking-induced conversion |
| TSE | = | transmissible spongiform encephalopathy |
| V | = | valine |
| vCJD | = | variant Creutzfeldt–Jakob disease |
| VPSPr | = | variably protease-sensitive prionopathy |
Introduction
Transmissible spongiform encephalopathies (TSE) or prion diseases are a unique group of fatal neurodegenerative diseases occurring in humans and mammals. Prion diseases can be sporadic, heritable, or acquired, they can be transmitted both naturally and experimentally, and as yet, there is no known cure. In 1996, an acquired human prion disease, variant Creutzfeldt–Jakob disease (vCJD), was described in the United Kingdom (UK) leading to a flurry of news reports, changes in government policies regarding the beef industry, a ban on exports of meat, restrictions on blood donations and a widespread fear that anyone could be infected. Since that first report, researchers and health professionals have endeavored to try and understand the disease, identify the infectious agent, assess transmission risks and ultimately improve diagnosis and find a cure. This review will summarize 18 y of research from identification of disease strain, epidemiology, and genetics, to assessing risks of transmission, diagnosis, and therapeutics, and finally the current issues of subclinical disease and ongoing surveillance.
BSE and vCJD: The Same Strain of Agent
In 1985, a novel neurodegenerative disease of cattle was recognized in the UK. Pathological examination of the brain material from these cattle suggested that this was a new TSE subsequently named bovine spongiform encephalopathy (BSE).Citation1 A further examination of these cases was performed following transmission of brain material to a panel of wild-type mice. The RIII, C57BL, and VM mice gave similar incubation periods, rankings, and vacuolation profiles for each isolate. Vacuolation distribution in the Prnp-a mice (RIII and C57BL) show distinctive profiles with higher levels or “peak” of vacuolation in the dorsal medulla, hypothalamus, and septum () whereas Prnp-b mice (VM) show peaks in the dorsal medulla, superior colliculus, thalamus, and septum.Citation2 This BSE signature was confirmed in a number of cases of BSE and was observed in a number of similar transmissions from cats,Citation3 kudu, and nyala.Citation4 Thus confirming a single agent was responsible for these new TSE cases in each species. Furthermore this agent was experimentally transmitted to sheep and goats.Citation5
Figure 1. Vacuolation scoring in the mouse brain. Lesion profile comparison of vCJD and BSE following transmission to RIII mice. Data shows mean lesion profile ± standard error of the mean (n ≥ 6). G1-G9, gray matter scoring regions; (G1) dorsal medulla, (G2) cerebellar cortex, (G3) superior colliculus, (G4) hypothalamus, (G5) thalamus, (G6) hippocampus, (G7) septum, (G8) retrosplenial and adjacent motor cortex, (G9) cingulate and adjacent motor cortex.
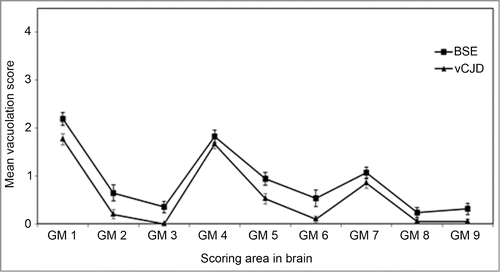
Ten years after BSE was recognized, the first case of an atypical form of Creutzfeldt–Jakob disease, termed “new variant CJD” (vCJD) in humans was identified;Citation6 both diseases were recognized to be prion diseases, raising serious concerns that BSE had now spread to humans via consumption of infected meat products.Citation7 Following the same protocols that had been used for the animal transmissions, a series of vCJD transmissions were set up. Initial results published in 1997 using RIII mice indicated that vCJD was indeed caused by the BSE agent.Citation8 At the same time, Hill et al.Citation9 showed similar results using FVB mice. Further studies have shown that incubation period rankings, lesion profiles and abnormal prion protein (PrPSc) deposition patterns are all identical to the BSE agent ( and ).Citation10,11 The definitive evidence that BSE and vCJD were the same strain came with the characterization of primary and secondary transmission of 10 cases of vCJD to mice. In this study both CNS and peripheral material was transmitted and showed that in all cases transmission characteristics were similar to BSE.Citation12 To date, all cases of UK 129MM (methionine homozygous) vCJD that have been characterized have shown similar strain characteristics to BSE.
Figure 2. Abnormal PrP deposition in hippocampus of RIII and VM mice following inoculation with BSE or vCJD. (A) BSE in RIII mouse; (B) vCJD in RIII mouse; (C) BSE in VM mouse; (D) vCJD in VM mouse (bar, 100 μm; anti-PrP antibody: 6H4).
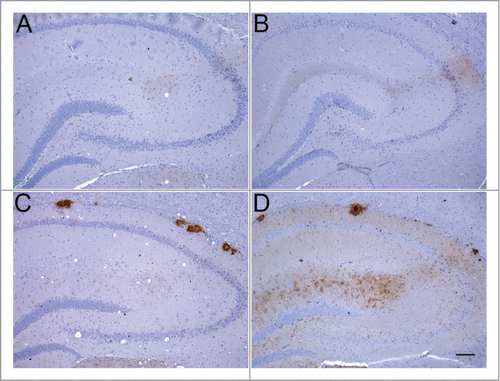
In addition to epidemiological, neuropathological, and biological evidence, the biochemical analysis of PrPSc deposited in both the BSE brain and vCJD brain also supports a link between the agents responsible. Western blotting of partially protease-resistant prion protein provides a surrogate for conformation and/or aggregation state and also reflects glycosylation site occupancy in the form of (generally 3) bands of protease-resistant prion protein (PrPres) of defined mobility (determined by N-terminal truncation) and relative abundance (determined by glycosylation). Using this method BSE can be differentiated from most forms of sheep scrapie.Citation13 Similarly vCJD can be distinguished from other human prion diseases, in particular sporadic CJD (sCJD),Citation14,15 whereas BSE and vCJD share both mobility type and have a similar glycoform ratio.Citation14 This BSE/vCJD PrPres type (referred to a type 2B or type 4) appears to be closely associated with the agent since (1) it also characterizes the PrPres that accumulates in peripheral tissues in clinical vCJD,Citation16 (2) it is maintained following secondary transmission of vCJD by blood transfusion both in the brain of clinical cases,Citation17 and in the spleen of asymptomatic or preclinical individuals,Citation18,19 (3) it is largely stable on transmission to wild-type and humanised transgenic miceCitation12,20 and (4) it is maintained in cell-free conversion systems in which either BSE or vCJD brain homogenates are used to seed conversion of normal human prion protein.Citation21
While type 2B PrPres provides a convenient additional diagnostic tool for human BSE identification,Citation22 it does not provide a complete description of PrPSc in cases of vCJD, nor does it provide a biochemical definition of the agent. The largest amount of PrPSc in the vCJD brain is actually protease sensitiveCitation23 and therefore does not figure in conventional PrPres typing. Even within the protease-resistant fraction of vCJD PrPSc there is evidence of a minority PrPres type.Citation24 The use of assays that do not depend upon protease-resistance as a definition of PrPSc show that aggregation state and stability are additional biochemical parameters that may be relevant to neurotoxicity and agent replication.Citation25,26
vCJD in the UK and Beyond
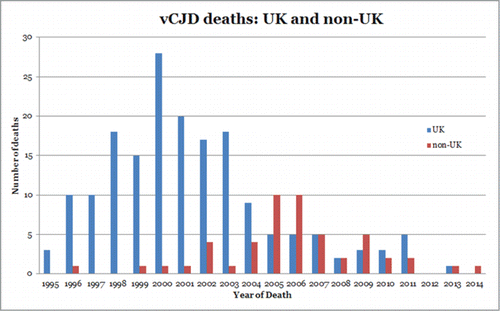
Since the first recorded case of vCJD in 1996, 177 cases of definite or probable vCJD have been reported in the UK (as of April 2014). The annual number of deaths reached a peak in 2000 with 28 deaths but since 2006, deaths from vCJD have levelled off at 2–5 per year with none reported in 2012 and only one in 2013Citation27 (). Originally restricted to the UK, 51 cases have now been reported in 11 other countries with a worldwide total of 228 (). Outside of the UK, most cases have occurred in France (27 cases to date) and are thought to be related to the peak in volume of beef imports originating from the UK during 1985–1995.Citation28 This potential relationship is shown in the peak in number of deaths from vCJD in France in 2005, 5 y after a similar peak in the UK.Citation28,29 Further evidence of the link between UK and French vCJD has arisen from comparative studies comparing epidemiologic, clinical, pathological, and biochemical analyses of vCJD cases from both countries indicating that the same strain of agent could be responsible.Citation30 The type 2B PrPres that characterizes UK vCJD casesCitation15 is also present in vCJD patients from FranceCitation30 and cases from Holland, Portugal, Spain, and Italy (Head and Ironside, unpublished information), consistent with the same strain of agent being involved in these different countries.
Figure 3. Reported incidence of vCJD deaths in the UK and in non-UK countries.
Exports of UK meat or cattle are assumed to have played a major role in the incidence of vCJD cases in other countries;Citation31 however there is the possibility that indigenous BSE or another strain of agent is responsible. In order to assess whether the same strain of agent is responsible for all vCJD cases worldwide, Diack et al.Citation32 performed strain typing of French, Italian, Dutch, and American cases of vCJD. These were all of the 129MM genotype and with limited exposure to UK BSE. Analysis of the transmission properties showed that the non-UK cases shared the same characteristics as UK cases of vCJD, however small differences were apparent in the incubation period rankings which are currently being studied.Citation32 The similar characteristics between UK and non-UK cases of vCJD suggest that current diagnostic criteria are sufficient to detect cases in all countries at this time. However these studies characterized “typical” cases of vCJD and do not take into account atypical cases or those occurring in genotypes other than 129MM.
All Codon 129 Genotypes are Susceptible to vCJD
Mutations and polymorphisms in the prion protein gene (PRNP) can influence or be associated with disease, i.e., E200K-129M in genetic CJD or A117V-129V in Gerstman–Sträussler–Scheinker disease (GSS). The codon 129 polymorphism (methionine (M)-valine (V)) of PRNP is known to be associated with susceptibility to CJDCitation33 with evidence from studies of kuru suggesting that heterozygosity is associated with increased survival times.Citation34 As stated, all definite and probable cases of clinical vCJD have been of the 129MM genotype which is in contrast to the normal distribution of genotypes in the general UK population; 42% 129MM, 47% 129MV, and 11% 129VVCitation35 and is suggestive of an association between vCJD susceptibility and genotype.
Experimental transmission studies have utilized gene targeted mice expressing human PrPC at physiological levels and overexpression models carrying each of the codon 129 genotypes to reveal that human-to-human transmission of vCJD is possible and that all genotypes have the potential to be affected.Citation9,20,36 Bishop et al.Citation20 used gene targeted models allowing direct comparison between mouse lines; these studies showed that transmission efficiency varied in the order MM > MV > VV with different pathological characteristics for each genotype. Mice expressing 129MM (HuMM) showed the greatest transmission efficiency and the earliest onset of both clinical disease and TSE related pathology. Although fewer HuMV mice were clinically affected and showed an extended incubation period, similar numbers demonstrated evidence of PrPSc compared with HuMM mice. In contrast only one HuVV mouse showed evidence of PrPSc.Citation20 This pattern of susceptibility has been repeated in a series of vCJD transmissions both from UK and non-UK material. This data suggests that in humans not only do all genotypes have the potential to be affected but that the different genotypes may manifest disease in different ways and indeed 129MV and 129VV individuals may have long asymptomatic incubation periods.
The evidence from the mouse studies has been shown in humans by the discovery of PrPSc in an asymptomatic PrP codon 129 heterozygote individual who died of a non-neurological disorder 5 y after receiving blood from an individual who later went onto develop clinical vCJD. In this individual evidence of PrPSc was found in the spleen and a cervical lymph node.Citation19 Transmission studies have now shown the spleen from this individual to be infectious.Citation37 A possible case of vCJD in a 129MV individual was reported in 2009, however vCJD was not confirmed since no autopsy was undertaken.Citation38 Additionally, retrospective studies of anonymised tonsil and appendix samples have shown evidence of PrPSc in all 3 genotypes giving further support to the evidence that all genotypes are susceptible to vCJD.Citation39,40
Modeling human genetic susceptibility to BSE using cell-free assays confirms the importance of methionine at codon 129 of the PRNP gene as a susceptibility factor, and shows the conversion efficiency to be MM > MV > VV, irrespective of whether the brain homogenate used to seed the reaction is vCJD, cattle BSE or experimental sheep BSE.Citation21
Blood as a Route of Transmission of vCJD
The UK shows the highest incidence of vCJD in the world.Citation27 At early stages of the epidemic, it was largely accepted that there was a minimal risk of transmission of vCJD from donations of peripheral blood/tissue from affected individuals to others via iatrogenic routes. That said great efforts were made to trace and track the fate of blood components used for transfusion from donors known to have vCJD.Citation41 Concomitantly, the likelihood of transmission of prion infection through blood, either by inoculation or transfusion and the distribution of prion-associated infectivity in blood components was being assessed using a range of animal models, typically small animal models.Citation42-48 These produced estimates of ∼10 infectious doses (ID)/ml in hamster blood.Citation47,49 Rodent models do not however represent accurately the procedures used in a clinical setting for blood transfusion. Large animal models such as sheep or deer infected with BSE or scrapie provide a suitable alternative to better assess the likelihood of transmission of prion disease following exposure by blood transfusion.Citation50-52 Larger volumes of blood can be collected from such animals and processed into components with similar specifications as those used for transfusion to humans.Citation53 Furthermore the peripheral pathogenesis of scrapie and BSE in sheep closely resembles that of humans affected with vCJD.Citation54,55 Over a decade ago, transfusion studies in sheep first demonstrated that all clinically-relevant blood components collected at both preclinical and clinical time points contained sufficient titers of disease-associated infectivity and could be transmitted to recipients after a single transfusion event. Moreover, the number of recipients that developed disease was suggestive that blood transfusion was a highly efficient route by which prion diseases could be transmitted.Citation56 Of note, in this and other studies, was the finding that the process of leucoreduction alone did not prevent the transmission of prion disease following blood transfusion.Citation49,53 The relevance of the latter point being that all components used for blood transfusion in humans are subject to universal leucoreduction. Data showing that blood from prion-infected animals was infectious was confirmed by other research groups using sheepCitation57,58 and deer blood transfusion models.Citation59-61
Since the late 1990s a number of risk reduction strategies were implemented to safeguard the UK blood (and blood product) supply. This included donor deferral and exclusion, importation of plasma from the USA for the preparation of plasma derivatives, i.e., clotting factors; the use of disposal instruments for certain surgical and dental procedures; and universal leucoreduction of all components used for transfusion.Citation62 Following a further risk assessment initiated by the Department of Health, selected groups of patients were informed that they could be considered to have a “small increased risk of carrying the vCJD agent” following receipt of certain batches of plasma products. These groups included hemophiliacs and those affected with other bleeding disorders and those with primary or secondary immunodeficiencies.Citation62,63
It was not until 2004 that blood from vCJD-infected humans was shown to pose a significant risk of acquiring prion infection. This followed the identification of 2 potential cases of blood-transfusion acquired vCJD.Citation19,64 A few years later saw the identification of another case of apparent transfusion-acquired vCJD.Citation65 These data were collated and cases presented in detail in 2006.Citation41,66 A fourth occurrence of transfusion-acquired vCJD was subsequently identified and all cases have been summarized in a recent review.Citation67 The 4 affected individuals were from the UK and all received non-leucoreduced red cell concentrates from UK donors, who were asymptomatic of infection at the time of donation but later died from vCJD. The transfusions took place between 1996 and 1999. Of the 4 transfusion recipients, 3 developed a clinical infection consistent with previously identified cases of vCJD (i.e., diseased-associated prion protein was evident in brain and peripheral lymphoid tissues examined post-mortem).Citation17 These individuals were identified as being methionine homozygous at codon 129 in the PRNP gene. The remaining transfusion recipient showed no clinical signs associated with vCJD or other neurological-type conditions and there was no evidence of disease-associated prion protein in the individual's brain and indeed the patient died of causes unrelated to vCJD.Citation19 Disease-associated prion protein was identified in selected lymphoid tissues such as the spleen and a cervical lymph node. Unlike the 3 clinical cases previously reported, this recipient was identified as having a different PRNP genotype being heterozygous (MV) at codon 129.
A surveillance program (established by the National CJD Research and Surveillance Unit and the UK Hemophilia Centre Doctors Organisation) identified the first case of vCJD infection in a hemophiliac patient.Citation18,68,69 The study examined biopsy and autopsy samples of lymphoid or brain tissue from a small numbers of samples submitted for investigation. The individual was an elderly male who resided in the UK. In conjunction with surgical procedures, the patient received numerous units of non-leucoreduced red cells and thousands of units of Factor VIII. The factor VIII was prepared from UK plasma pools and it was found that some of the pooled-plasma could be traced back to a donor who died from vCJD. The individual showed no signs of vCJD or other neurological conditions and was MV at codon 129 in the PRNP gene. Upon repeated examination, a specific area of spleen was positive for the abnormal form of the prion protein. A subsequent risk assessment found that of all possible sources of vCJD infection, including dietary exposure, the mostly likely was determined as treatment with UK-sourced clotting factors.Citation70 To date, there have been no further cases of vCJD acquired following the transfusion of blood, blood components or clotting factors. While there has been no documented evidence, to date, of the transmission of sCJD infection following blood transfusion in humans,Citation41 a recent, though limited study, has reported the presence of disease-associated infectivity in plasma obtained from 2 patients affected with sCJD.Citation71
Although estimates of the infectious titer of blood from patients with vCJD are lowCitation71,72 it has been demonstrated that blood and components from asymptomatic individuals appear capable of transmitting vCJD-infection following blood transfusion. Major efforts have been made toward the development of screening assays and diagnostic tests for vCJD in bloodCitation72-75; the development and implementation of prion reduction filtersCitation76-80; understanding the numbers of individuals who may be sub-clinically affected with vCJDCitation39,Citation81-84 and what this really means in terms of further spread of vCJD. There are significant challenges to be faced in each of these areas, which are further confounded by the absence of an available treatment for vCJD.
Prevalence of Asymptomatic vCJD Infection
The UK population had a wide exposure to the BSE agent through contaminated meat products in the food chain in the 1980s and early 1990s, resulting in 177 definite cases of vCJD to date. Of the cases genotyped, all were methionine homozygotes at codon 129 in the PRNP gene. However there are ongoing concerns over vCJD infection in other codon 129 genotypes with potentially longer incubation periods. This has prompted a series of tissue–based studies on the prevalence of vCJD infection in lymphoid tissues (appendices and tonsils) removed surgically as part of treatment for appendicitis, tonsillitis, and related disorders in otherwise healthy individuals with no neurological symptoms. Variant CJD differs from other human prion diseases in the widespread involvement of lymphoid tissues by the causative agent which is detectable in follicular dendritic cells and is associated with infectivity.Citation16,85
Review of paraffin-embedded appendices that had been resected from a small number of individuals before the onset of vCJD revealed that prion protein was detectable in the lymphoid follicles in the wall of the appendix for at least 2 y prior to the onset of vCJD symptoms.Citation86 This observation allowed the possibility of a large-scale retrospective survey of appendix and tonsil tissues from histopathology departments across the UK to determine the extent of asymptomatic vCJD infection as revealed by immunohistochemistry on paraffin-embedded tissues. These studies have proven challenging in terms of logistics and ethics and have proceeded on the basis of using anonymised specimens that are not directly linkable to any individual. The first of these studies reported in 2004 an estimated prevalence of asymptomatic vCJD infection in 237 per million in 4000 individuals in the UK (3 in 12 674 positive specimens tested), but with very wide 95% confidence intervals (49–692 per million).Citation83 Two subsequent prospective studies on tonsil tissues collected frozen tissue samples as well as paraffin-embedded tissues, which allowed the use of enzyme immunoassays and western blotting in addition to immunohistochemistry for the detection of the abnormal prion protein.Citation81,82,87 No positives were detected in the frozen tissue samples from either study (2000 in Frosh et al.Citation87; 32 661 in Clewley et al,Citation81). Immunohistochemistry was subsequently performed on 10 075 samples from the de Marco et al.Citation82 study, with 1 apparent positive detected.
In order to resolve the findings from these studies, a larger unlinked and anonymised immunohistochemical survey was performed on archived paraffin-embedded appendix samples from 41 histopathology departments in the UK.Citation39 Of the 32 441 samples assessed, 16 were positive for abnormal prion protein, giving an overall prevalence of 493 per million (95% confidence intervals 282–801 per million), which is broadly in keeping with the results of the earlier study by Hilton et al.Citation83 PRNP codon 129 genotype analysis of the positive cases showed that all possible genotypes were involved, with a predominance of the valine homozygous genotype,Citation39 as for the Hilton et al. study,Citation40 and in contrast with the definite cases of vCJD identified to date. These findings have a wide range of implications, including the need for continuing surveillance of human prion diseases in the UK and the risks of secondary vCJD transmission from asymptomatic infected individuals via surgical instruments or blood transfusion; the latter is now the subject of a UK Parliamentary Inquiry (Parliamentary Select Committee on Science and Technology, 2013Citation88).
Diagnostics and Treatment
The diagnosis of vCJD rests on recognizing the typical phenotype and applying appropriate specialist investigations, in particular MRI brain scan (). The clinical features are remarkably stereotyped. There is an initial phase of around 6 mo dominated by psychiatric symptoms, including depression, delusions, and anxietyCitation89 followed by the rapid development of neurological features,Citation90 typically confusion, ataxia, and involuntary movements, which may be choreiform, dystonic, or myoclonic. The duration of illness from onset to death averages 14 moCitation91 in contrast to sCJD in which the mean survival is 4 mo.
Figure 4. MRI brain scan in variant CJD. FLAIR axial section at the level of the basal ganglia showing bilateral symmetrical dorsomedial and pulvinar thalamic hyperintensity. Courtesy of Dr David Summers.
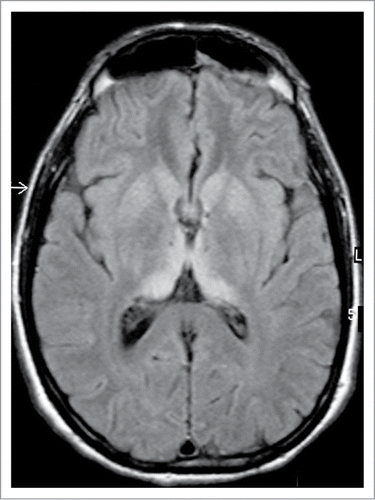
The electroencephalogram does not show the periodic sharp wave complexes that are seen in sCJD, except rarely in the terminal stages of the illnessCitation92 and the CSF 14–3-3 immunoassay is only positive in about half the cases.Citation93 The CSF RT-QuIC has been negative in vCJD in all assays to date. The most helpful investigation is MRI brain scan, which shows high signal in the pulvinar region of the thalamus, the so-called hockey stick sign, on FLAIR () and DWI sequences in over 90% of cases.Citation94 Tonsil biopsy shows immunostaining and deposition of type 2B or type 4 PrPres in the majority of cases,Citation95,96 but this test is invasive, and definitive diagnosis rests on neuropathological examination of brain tissue, usually at post-mortem.
Highly sensitive and specific diagnostic criteria, including a combination of core clinical features and the results of MRI brain scan and pathology, have been formulatedCitation97 and validated.Citation98 Cases classified as definite or probable are reported by international surveillance systems as the likelihood of accurate diagnosis in possible cases is uncertain.
The phenotype in cases of vCJD in an MV or VV genetic background cannot be predicted and continued vigilance is necessary in order to identify such cases.
Treatment of vCJD has been attempted using a range of medications, but none have been proven to be effective. Initial reports of improvement following treatment with quinacrine have not been confirmed in observational trialsCitation99,100 and this drug is no longer used in vCJD treatment. Studies in animal models raised the possibility that pentosan polysulphate (PPS) might be a candidate treatment for vCJDCitation101 and extended survival has been reported in a small number of treated cases.Citation102,103 However, this medication has to be given by intraventricular infusion, requiring a neurosurgical procedure, and treated patients continued to decline with no reversal of severe neurological deficits.Citation104 Post-mortem examination of one case of vCJD treated with PPS showed extensive and severe pathology.Citation105 Some cases of vCJD received doxycycline with no obvious benefit and a controlled trial in sCJD has not demonstrated efficacy.Citation99
Emergence of Novel Strains
Identification of novel strains involves veterinary and medical vigilance, but it also requires a proper and full characterization of known prion agents. While the deployment of wild-type mouse panels, transgenic mice, and non-human primates all rapidly concluded that vCJD was a novel human prion strain related to BSE (see above), determining how many distinct human prion strains there are has proved surprisingly difficult, especially for sCJD. Transmission studies in humanised transgenic mice and non-human primates point to 4 major groups within sporadic, iatrogenic CJD, Kuru, and some genetic CJD cases, termed M1, V1, M2, and V2.Citation22,106 Sporadic fatal insomnia and fatal familial insomnia (FFI) together may represent a sixth strainCitation107 and 2 further transmissible phenotypes can be derived from GSS disease: one involving a transmissible amyloid phenotype, the other a fully transmissible spongiform encephalopathy.Citation108,109 The transmission properties of PrP cerebral amyloid angiopathy and Variably ProteaseSensitive Prionopathy (VPSPr) remain to be reported. The relationship between human disease phenotypes, agent strain, and prion biochemistry is further complicated by the now widely recognized phenomenon of distinct PrPres type co-occurrence in the sCJD, vCJD and VPSPr brain.Citation15,24,110
Surveillance for BSE in cattle, sheep, and goats has identified new (or newly discovered) animal prion diseases including atypical scrapie in sheep and so called H- and L-type BSE in cattle. These along with chronic wasting disease (CWD) in deer and elk represent a potential zoonotic risk to human health that is hard to quantify. While Wilson et al.Citation111 have shown no transmission of CWD, BASE, H-type BSE, and atypical scrapie to mice expressing wild-type levels of human PrP, Kong et al.Citation112 demonstrated transmission of BASE to an alternative line of mice expressing wild-type levels of human PrP. In contrast, Beringue et al.Citation113 showed transmission of BASE to mice overexpressing human PrP but no evidence of H-type BSE transmission, furthermore no evidence of CWD transmission to overexpressing mice has been identified.Citation114,115 This difference in transmission results may be due to different genetic backgrounds or differences in PrP expression levels between the different mouse lines. An alternative to modeling the species barrier is the cell-free conversion assay which points to CWD as the animal prion disease with the greatest zoonotic potential, after (and very much less than) BSE.Citation116
Surveillance
Continued surveillance for long-term effects of BSE exposure in the UK human population appears necessary for the foreseeable future in order to discount possible second wave epidemics that might depend on genetic susceptibility, subclinical infection, and secondary transmission or disease in defined “at risk” patient groups such as hemophiliacs or patient groups in which full ascertainment is difficult, such as the elderly.
However, an additional concern is associated with idiopathic human prion disease. Sporadic CJD is not a uniform condition and the phenotype is clearly influenced by the codon 129 genotype of the patient and the prion protein type that accumulates in their brain. The etiological basis of the condition might be presumed to be spontaneously occurring, but this is not known with certainty in general, or in individual specific cases. Neither are the molecular mechanisms of spontaneous conversion of the prion protein to its pathogenic form well understood or easily investigated. Additionally, surveillance identifies apparently sporadic cases of human prion disease that do not fit well into currently accepted classification systems. This is exemplified by the recent identification of a new human prion disease (VPSPr by Gambetti et al.Citation117) and its prospective and retrospective identification in other countries subsequently.Citation118,110 The true prevalence, the relationship to sCJD and the risk to public health of VPSPr are yet to be determined.
Conclusions
Since the identification of vCJD we have made progress in identifying routes of infection, controlling further infection, producing models of disease, developing decontamination procedures, and understanding susceptibility to disease. The vCJD epidemic in the UK now appears to be in decline and it appears that the control measures in food production and blood supplies have prevented further vCJD cases arising through dietary/infected blood exposure.
Despite this, there are still ongoing concerns over cases of vCJD arising in countries where little or no exposure to UK meat products have occurred, the presence of subclinical vCJD in the UK population with the possibility of further human-to-human transmission and the identification of new strains of human prion disease. These scenarios necessitate ongoing studies in understanding transmission properties, disease diagnosis, and therapeutics. The identification of novel human prion diseases and the current estimates of subclinical vCJD infections show the importance of continued CJD surveillance and maintaining control measures already in place to protect human health.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We are grateful to our colleagues at The Roslin Institute, the National CJD Research and Surveillance Unit, and the Edinburgh Brain Bank for help in these studies.
Funding
The work described here has been supported by BBSRC, Medical Research Council, and Department of Health. This report is independent research commissioned and funded by the Department of Health, Policy Research Programme. The views expressed in this publication are those of the authors and not necessarily those of the Department of Health.
References
- Wells GA, Scott AC, Johnson CT, Gunning RF, Hancock RD, Jeffrey M, Dawson M, Bradley R. A novel progressive spongiform encephalopathy in cattle. Vet Rec 1987; 121:419-20; PMID:3424605; http://dx.doi.org/10.1136/vr.121.18.419
- Fraser H, Bruce ME, Chree A, McConnell I, Wells GA. Transmission of bovine spongiform encephalopathy and scrapie to mice. J Gen Virol 1992; 73:1891-7; PMID:1645134; http://dx.doi.org/10.1099/0022-1317-73-8-1891
- Fraser H, Pearson GR, McConnell I, Bruce ME, Wyatt JM, Gruffydd-Jones TJ. Transmission of feline spongiform encephalopathy to mice. Vet Rec 1994; 134:449; PMID:8048218; http://dx.doi.org/10.1136/vr.134.17.449
- Bruce M, Chree A, McConnell I, Foster J, Pearson G, Fraser H. Transmission of bovine spongiform encephalopathy and scrapie to mice: strain variation and the species barrier. Philos Trans R Soc Lond B Biol Sci 1994; 343:405-11; PMID:7913758; http://dx.doi.org/10.1098/rstb.1994.0036
- Foster JD, Hope J, Fraser H. Transmission of bovine spongiform encephalopathy to sheep and goats. Vet Rec. 1993;133:339-41. PMID:8236676
- Will RG, Ironside JW, Zeidler M, Cousens SN, Estibeiro K, Alperovitch A, Poser S, Pocchiari M, Hofman A, Smith PG. A new variant of Creutzfeldt-Jakob disease in the UK. Lancet 1996; 347:921-5; PMID:8598754; http://dx.doi.org/10.1016/S0140-6736(96)91412-9
- Ward HJ, Everington D, Cousens SN, Smith-Bathgate B, Leitch M, Cooper S, Heath C, Knight RS, Smith PG, Will RG. Risk factors for variant Creutzfeldt-Jakob disease: a case-control study. Ann Neurol 2006; 59:111-20; PMID:16287153; http://dx.doi.org/10.1002/ana.20708
- Bruce ME, Will RG, Ironside JW, McConnell I, Drummond D, Suttie A, McCardle L, Chree A, Hope J, Birkett C, et al. Transmissions to mice indicate that ‘new variant’ CJD is caused by the BSE agent. Nature 1997; 389:498-501; PMID:9333239; http://dx.doi.org/10.1038/39057
- Hill AF, Desbruslais M, Joiner S, Sidle KC, Gowland I, Collinge J, Doey LJ, Lantos P. The same prion strain causes vCJD and BSE. Nature 1997; 389:448-50, 526; PMID:9333232; http://dx.doi.org/10.1038/38925
- Brown DA, Bruce ME, Fraser JR. Comparison of the neuropathological characteristics of bovine spongiform encephalopathy (BSE) and variant Creutzfeldt-Jakob disease (vCJD) in mice. Neuropathol Appl Neurobiol 2003; 29:262-72; PMID:12787323; http://dx.doi.org/10.1046/j.1365-2990.2003.00462.x
- Bruce ME. TSE strain variation. Br Med Bull 2003; 66:99-108; PMID:14522852; http://dx.doi.org/10.1093/bmb/66.1.99
- Ritchie DL, Boyle A, McConnell I, Head MW, Ironside JW, Bruce ME. Transmissions of variant Creutzfeldt-Jakob disease from brain and lymphoreticular tissue show uniform and conserved bovine spongiform encephalopathy-related phenotypic properties on primary and secondary passage in wild-type mice. J Gen Virol 2009; 90:3075-82; PMID:19656962; http://dx.doi.org/10.1099/vir.0.013227-0
- Stack MJ, Chaplin MJ, Clark J. Differentiation of prion protein glycoforms from naturally occurring sheep scrapie, sheep-passaged scrapie strains (CH1641 and SSBP1), bovine spongiform encephalopathy (BSE) cases and Romney and Cheviot breed sheep experimentally inoculated with BSE using two monoclonal antibodies. Acta Neuropathol 2002; 104:279-86; PMID:12172914
- Collinge J, Sidle KC, Meads J, Ironside J, Hill AF. Molecular analysis of prion strain variation and the aetiology of ‘new variant’ CJD. Nature 1996; 383:685-90; PMID:8878476; http://dx.doi.org/10.1038/383685a0
- Head MW, Bunn TJR, Bishop MT, McLoughlin V, Lowrie S, McKimmie CS, Williams MC, McCardle L, MacKenzie J, Knight R, et al. Prion protein heterogeneity in sporadic but not variant Creutzfeldt-Jakob disease: UK cases 1991-2002. Ann Neurol 2004; 55:851-9; PMID:15174020; http://dx.doi.org/10.1002/ana.20127
- Head MW, Ritchie D, Smith N, McLoughlin V, Nailon W, Samad S, Masson S, Bishop M, McCardle L, Ironside JW. Peripheral tissue involvement in sporadic, iatrogenic, and variant Creutzfeldt-Jakob disease: an immunohistochemical, quantitative, and biochemical study. Am J Pathol 2004; 164:143-53; PMID:14695328; http://dx.doi.org/10.1016/S0002-9440(10)63105-7
- Head MW, Yull HM, Ritchie DL, Bishop MT, Ironside JW. Pathological investigation of the first blood donor and recipient pair linked by transfusion-associated variant Creutzfeldt-Jakob disease transmission. Neuropathol Appl Neurobiol 2009; 35:433-6; PMID:19490428; http://dx.doi.org/10.1111/j.1365-2990.2009.01025.x
- Peden A, McCardle L, Head MW, Love S, Ward HJT, Cousens SN, Keeling DM, Millar CM, Hill FGH, Ironside JW. Variant CJD infection in the spleen of a neurologically asymptomatic UK adult patient with haemophilia. Haemophilia 2010; 16:296-304; PMID:20070383; http://dx.doi.org/10.1111/j.1365-2516.2009.02181.x
- Peden AH, Head MW, Ritchie DL, Bell JE, Ironside JW. Preclinical vCJD after blood transfusion in a PRNP codon 129 heterozygous patient. Lancet 2004; 364:527-9; PMID:15302196; http://dx.doi.org/10.1016/S0140-6736(04)16811-6
- Bishop MT, Hart P, Aitchison L, Baybutt HN, Plinston C, Thomson V, Tuzi NL, Head MW, Ironside JW, Will RG, et al. Predicting susceptibility and incubation time of human-to-human transmission of vCJD. Lancet Neurol 2006; 5:393-8; PMID:16632309; http://dx.doi.org/10.1016/S1474-4422(06)70413-6
- Jones M, Wight D, Barron R, Jeffrey M, Manson J, Prowse C, Ironside JW, Head MW. Molecular model of prion transmission to humans. Emerg Infect Dis 2009; 15:2013-6; PMID:19961689; http://dx.doi.org/10.3201/eid1512.090194
- Parchi P, Notari S, Weber P, Schimmel H, Budka H, Ferrer I, Haik S, Hauw JJ, Head MW, Ironside JW, et al. Inter-laboratory assessment of PrPSc typing in creutzfeldt-jakob disease: a Western blot study within the NeuroPrion Consortium. Brain Pathol 2009; 19:384-91; PMID:18624793; http://dx.doi.org/10.1111/j.1750-3639.2008.00187.x
- Choi YP, Gröner A, Ironside JW, Head MW. Comparison of the level, distribution and form of disease-associated prion protein in variant and sporadic Creutzfeldt-Jakob diseased brain using conformation-dependent immunoassay and Western blot. J Gen Virol 2011; 92:727-32; PMID:21123539; http://dx.doi.org/10.1099/vir.0.026948-0
- Yull HM, Ritchie DL, Langeveld JP, van Zijderveld FG, Bruce ME, Ironside JW, Head MW. Detection of type 1 prion protein in variant Creutzfeldt-Jakob disease. Am J Pathol 2006; 168:151-7; PMID:16400018; http://dx.doi.org/10.2353/ajpath.2006.050766
- Choi YP, Gröner A, Ironside JW, Head MW. Correlation of polydispersed prion protein and characteristic pathology in the thalamus in variant Creutzfeldt-Jakob disease: implication of small oligomeric species. Brain Pathol 2011; 21:298-307; PMID:21029243; http://dx.doi.org/10.1111/j.1750-3639.2010.00446.x
- Choi YP, Peden AH, Gröner A, Ironside JW, Head MW. Distinct stability states of disease-associated human prion protein identified by conformation-dependent immunoassay. J Virol 2010; 84:12030-8; PMID:20844046; http://dx.doi.org/10.1128/JVI.01057-10
- National CJD Research & Surveillance Unit Data and Reports. Latest NCJDSRU CJD Monthly Statistics. [cited 2014 April]. Available from: http://www.cjd.ed.ac.uk/documents/figs.pdf.
- Chadeau-Hyam M, Alpérovitch A. Risk of variant Creutzfeldt-Jakob disease in France. Int J Epidemiol 2005; 34:46-52; PMID:15649960; http://dx.doi.org/10.1093/ije/dyh374
- Brandel JP, Peckeu L, Haïk S. The French surveillance network of Creutzfeldt-Jakob disease. Epidemiological data in France and worldwide. Transfus Clin Biol 2013; 20:395-7; PMID:23587616; http://dx.doi.org/10.1016/j.tracli.2013.02.029
- Brandel JP, Heath CA, Head MW, Levavasseur E, Knight R, Laplanche JL, Langeveld JP, Ironside JW, Hauw JJ, Mackenzie J, et al. Variant Creutzfeldt-Jakob disease in France and the United Kingdom: Evidence for the same agent strain. Ann Neurol 2009; 65:249-56; PMID:19334063; http://dx.doi.org/10.1002/ana.21583
- Sanchez-Juan P, Cousens SN, Will RG, van Duijn CM. Source of variant Creutzfeldt-Jakob disease outside United Kingdom. Emerg Infect Dis 2007; 13:1166-9; PMID:17953086; http://dx.doi.org/10.3201/eid1308.070178
- Diack AB, Ritchie D, Bishop M, Pinion V, Brandel JP, Haik S, Tagliavini F, Van Duijn C, Belay ED, Gambetti P, et al. Constant transmission properties of variant Creutzfeldt-Jakob disease in 5 countries. Emerg Infect Dis 2012; 18:1574-9; PMID:23017202; http://dx.doi.org/10.3201/eid1810.120792
- Pocchiari M, Puopolo M, Croes EA, Budka H, Gelpi E, Collins S, Lewis V, Sutcliffe T, Guilivi A, Delasnerie-Laupretre N, et al. Predictors of survival in sporadic Creutzfeldt-Jakob disease and other human transmissible spongiform encephalopathies. Brain 2004; 127:2348-59; PMID:15361416; http://dx.doi.org/10.1093/brain/awh249
- Lee HS, Brown P, Cervenáková L, Garruto RM, Alpers MP, Gajdusek DC, Goldfarb LG. Increased susceptibility to Kuru of carriers of the PRNP 129 methionine/methionine genotype. J Infect Dis 2001; 183:192-6; PMID:11120925; http://dx.doi.org/10.1086/317935
- Nurmi MH, Bishop M, Strain L, Brett F, McGuigan C, Hutchison M, Farrell M, Tilvis R, Erkkilä S, Simell O, et al. The normal population distribution of PRNP codon 129 polymorphism. Acta Neurol Scand 2003; 108:374-8; PMID:14616310; http://dx.doi.org/10.1034/j.1600-0404.2003.00199.x
- Asante EA, Linehan JM, Desbruslais M, Joiner S, Gowland I, Wood AL, Welch J, Hill AF, Lloyd SE, Wadsworth JD, et al. BSE prions propagate as either variant CJD-like or sporadic CJD-like prion strains in transgenic mice expressing human prion protein. EMBO J 2002; 21:6358-66; PMID:12456643; http://dx.doi.org/10.1093/emboj/cdf653
- Bishop MT, Diack AB, Ritchie DL, Ironside JW, Will RG, Manson JC. Prion infectivity in the spleen of a PRNP heterozygous individual with subclinical variant Creutzfeldt-Jakob disease. Brain 2013; 136:1139-45; PMID:23449776; http://dx.doi.org/10.1093/brain/awt032
- Kaski D, Mead S, Hyare H, Cooper S, Jampana R, Overell J, Knight R, Collinge J, Rudge P. Variant CJD in an individual heterozygous for PRNP codon 129. Lancet 2009; 374:2128; PMID:20109837; http://dx.doi.org/10.1016/S0140-6736(09)61568-3
- Gill ON, Spencer Y, Richard-Loendt A, Kelly C, Dabaghian R, Boyes L, Linehan J, Simmons M, Webb P, Bellerby P, et al. Prevalent abnormal prion protein in human appendixes after bovine spongiform encephalopathy epizootic: large scale survey. BMJ 2013; 347:f5675; PMID:24129059; http://dx.doi.org/10.1136/bmj.f5675
- Ironside JW, Bishop MT, Connolly K, Hegazy D, Lowrie S, Le Grice M, Ritchie DL, McCardle LM, Hilton DA. Variant Creutzfeldt-Jakob disease: prion protein genotype analysis of positive appendix tissue samples from a retrospective prevalence study. BMJ 2006; 332:1186-8; PMID:16606639; http://dx.doi.org/10.1136/bmj.38804.511644.55
- Hewitt PE, Llewelyn CA, Mackenzie J, Will RG. Creutzfeldt-Jakob disease and blood transfusion: results of the UK Transfusion Medicine Epidemiological Review study. Vox Sang 2006; 91:221-30; PMID:16958834; http://dx.doi.org/10.1111/j.1423-0410.2006.00833.x
- Bons N, Lehmann S, Mestre-Francès N, Dormont D, Brown P. Brain and buffy coat transmission of bovine spongiform encephalopathy to the primate Microcebus murinus. Transfusion 2002; 42:513-6; PMID:12084158; http://dx.doi.org/10.1046/j.1537-2995.2002.00098.x
- Brown P, Rohwer RG, Dunstan BC, MacAuley C, Gajdusek DC, Drohan WN. The distribution of infectivity in blood components and plasma derivatives in experimental models of transmissible spongiform encephalopathy. Transfusion 1998; 38:810-6; PMID:9738619; http://dx.doi.org/10.1046/j.1537-2995.1998.38998408999.x
- Brown P, Cervenáková L, McShane LM, Barber P, Rubenstein R, Drohan WN. Further studies of blood infectivity in an experimental model of transmissible spongiform encephalopathy, with an explanation of why blood components do not transmit Creutzfeldt-Jakob disease in humans. Transfusion 1999; 39:1169-78; PMID:10604242; http://dx.doi.org/10.1046/j.1537-2995.1999.39111169.x
- Cervenakova L, Yakovleva O, McKenzie C, Kolchinsky S, McShane L, Drohan WN, Brown P. Similar levels of infectivity in the blood of mice infected with human-derived vCJD and GSS strains of transmissible spongiform encephalopathy. Transfusion 2003; 43:1687-94; PMID:14641865; http://dx.doi.org/10.1046/j.0041-1132.2003.00586.x
- Herzog C, Salès N, Etchegaray N, Charbonnier A, Freire S, Dormont D, Deslys JP, Lasmézas CI. Tissue distribution of bovine spongiform encephalopathy agent in primates after intravenous or oral infection. Lancet 2004; 363:422-8; PMID:14962521; http://dx.doi.org/10.1016/S0140-6736(04)15487-1
- Holada K, Vostal JG, Theisen PW, MacAuley C, Gregori L, Rohwer RG. Scrapie infectivity in hamster blood is not associated with platelets. J Virol 2002; 76:4649-50; PMID:11932431; http://dx.doi.org/10.1128/JVI.76.9.4649-4650.2002
- Miekka SI, Forng RY, Rohwer RG, MacAuley C, Stafford RE, Flack SL, MacPhee M, Kent RS, Drohan WN. Inactivation of viral and prion pathogens by gamma-irradiation under conditions that maintain the integrity of human albumin. Vox Sang 2003; 84:36-44; PMID:12542732; http://dx.doi.org/10.1046/j.1423-0410.2003.00256.x
- Gregori L, McCombie N, Palmer D, Birch P, Sowemimo-Coker SO, Giulivi A, Rohwer RG. Effectiveness of leucoreduction for removal of infectivity of transmissible spongiform encephalopathies from blood. Lancet 2004; 364:529-31; PMID:15302197; http://dx.doi.org/10.1016/S0140-6736(04)16812-8
- Houston F, Foster JD, Chong A, Hunter N, Bostock CJ. Transmission of BSE by blood transfusion in sheep. Lancet 2000; 356:999-1000; PMID:11041403; http://dx.doi.org/10.1016/S0140-6736(00)02719-7
- Hunter N, Foster J, Chong A, McCutcheon S, Parnham D, Eaton S, MacKenzie C, Houston F. Transmission of prion diseases by blood transfusion. J Gen Virol 2002; 83:2897-905; PMID:12388826
- Hunter N, Houston F. Can prion diseases be transmitted between individuals via blood transfusion: evidence from sheep experiments. Dev Biol (Basel) 2002; 108:93-8; PMID:12220147
- McCutcheon S, Alejo Blanco AR, Houston EF, de Wolf C, Tan BC, Smith A, Groschup MH, Hunter N, Hornsey VS, MacGregor IR, et al. All clinically-relevant blood components transmit prion disease following a single blood transfusion: a sheep model of vCJD. PLoS One 2011; 6:e23169; PMID:21858015; http://dx.doi.org/10.1371/journal.pone.0023169
- Ironside JW, McCardle L, Horsburgh A, Lim Z, Head MW. Pathological diagnosis of variant Creutzfeldt-Jakob disease. APMIS 2002; 110:79-87; PMID:12064259; http://dx.doi.org/10.1034/j.1600-0463.2002.100110.x
- Foster JD, Parnham DW, Hunter N, Bruce M. Distribution of the prion protein in sheep terminally affected with BSE following experimental oral transmission. J Gen Virol 2001; 82:2319-26; PMID:11562525
- Houston F, McCutcheon S, Goldmann W, Chong A, Foster J, Sisó S, González L, Jeffrey M, Hunter N. Prion diseases are efficiently transmitted by blood transfusion in sheep. Blood 2008; 112:4739-45; PMID:18647958; http://dx.doi.org/10.1182/blood-2008-04-152520
- Andréoletti O, Litaise C, Simmons H, Corbière F, Lugan S, Costes P, Schelcher F, Vilette D, Grassi J, Lacroux C. Highly efficient prion transmission by blood transfusion. PLoS Pathog 2012; 8:e1002782; PMID:22737075; http://dx.doi.org/10.1371/journal.ppat.1002782
- Lacroux C, Vilette D, Fernández-Borges N, Litaise C, Lugan S, Morel N, Corbière F, Simon S, Simmons H, Costes P, et al. Prionemia and leukocyte-platelet-associated infectivity in sheep transmissible spongiform encephalopathy models. J Virol 2012; 86:2056-66; PMID:22156536; http://dx.doi.org/10.1128/JVI.06532-11
- Mathiason CK, Hayes-Klug J, Hays SA, Powers J, Osborn DA, Dahmes SJ, Miller KV, Warren RJ, Mason GL, Telling GC, et al. B cells and platelets harbor prion infectivity in the blood of deer infected with chronic wasting disease. J Virol 2010; 84:5097-107; PMID:20219916; http://dx.doi.org/10.1128/JVI.02169-09
- Mathiason CK, Hays SA, Powers J, Hayes-Klug J, Langenberg J, Dahmes SJ, Osborn DA, Miller KV, Warren RJ, Mason GL, et al. Infectious prions in pre-clinical deer and transmission of chronic wasting disease solely by environmental exposure. PLoS One 2009; 4:e5916; PMID:19529769; http://dx.doi.org/10.1371/journal.pone.0005916
- Mathiason CK, Powers JG, Dahmes SJ, Osborn DA, Miller KV, Warren RJ, Mason GL, Hays SA, Hayes-Klug J, Seelig DM, et al. Infectious prions in the saliva and blood of deer with chronic wasting disease. Science 2006; 314:133-6; PMID:17023660; http://dx.doi.org/10.1126/science.1132661
- DH. vCJD: Further precautionary measures announced. http://webarchive.nationalarchives.gov.uk/+/www.dh.gov.uk/en/Publicationsandstatistics/Pressreleases/DH_4089689. 2004.
- HPA. Variant CJD and blood products. http://www.hpa.org.uk/webw/HPAweb&HPAwebStandard/HPAweb_C/1195733818681?p=1191942152861. 2004.
- Llewelyn CA, Hewitt PE, Knight RS, Amar K, Cousens S, Mackenzie J, Will RG. Possible transmission of variant Creutzfeldt-Jakob disease by blood transfusion. Lancet 2004; 363:417-21; PMID:14962520; http://dx.doi.org/10.1016/S0140-6736(04)15486-X
- Wroe SJ, Pal S, Siddique D, Hyare H, Macfarlane R, Joiner S, Linehan JM, Brandner S, Wadsworth JD, Hewitt P, et al. Clinical presentation and pre-mortem diagnosis of variant Creutzfeldt-Jakob disease associated with blood transfusion: a case report. Lancet 2006; 368:2061-7; PMID:17161728; http://dx.doi.org/10.1016/S0140-6736(06)69835-8
- Hewitt PE, Llewelyn CA, Mackenzie J, Will RG. Three reported cases of variant Creutzfeldt-Jakob disease transmission following transfusion of labile blood components. Vox Sang 2006; 91:348; PMID:17105612; http://dx.doi.org/10.1111/j.1423-0410.2006.00837.x
- Ironside JW. Variant Creutzfeldt-Jakob disease: an update. Folia Neuropathol 2012; 50:50-6; PMID:22505363
- Ironside JW, Head MW, Peden A, Ward H. Asymptomatic vCJD infection detected at autopsy in a UK haemophilic patient. Haemophilia 2010; 16:29.
- Millar CM, Connor N, Dolan G, Lee CA, Makris M, Wilde J, Winter M, Ironside JW, Gill N, Hill FG. Risk reduction strategies for variant Creutzfeldt-Jakob disease transmission by UK plasma products and their impact on patients with inherited bleeding disorders. Haemophilia 2010; 16:305-15; PMID:20487442; http://dx.doi.org/10.1111/j.1365-2516.2010.02220.x
- DH. vCJD risk assessment calculation for a patient with multiple routes of exposure. http://webarchive.nationalarchives.gov.uk/20130107105354/http://www.dh.gov.uk/en/Publicationsandstatistics/Publications/PublicationsPolicyAndGuidance/DH_100357., 2009.
- Douet JY, Zafar S, Perret-Liaudet A, Lacroux C, Lugan S, Aron N, Cassard H, Ponto C, Corbière F, Torres JM, et al. Detection of infectivity in blood of persons with variant and sporadic Creutzfeldt-Jakob disease. Emerg Infect Dis 2014; 20:114-7; PMID:24377668; http://dx.doi.org/10.3201/eid2001.130353
- Gregori L. A prototype assay to detect vCJD-infected blood. Lancet 2011; 377:444-6; PMID:21295340; http://dx.doi.org/10.1016/S0140-6736(11)60057-3
- Cooper JK, Andrews N, Ladhani K, Bujaki E, Minor PD. Evaluation of a test for its suitability in the diagnosis of variant Creutzfeldt-Jakob disease. Vox Sang 2013; 105:196-204; PMID:23772892; http://dx.doi.org/10.1111/vox.12037
- Edgeworth JA, Farmer M, Sicilia A, Tavares P, Beck J, Campbell T, Lowe J, Mead S, Rudge P, Collinge J, et al. Detection of prion infection in variant Creutzfeldt-Jakob disease: a blood-based assay. Lancet 2011; 377:487-93; PMID:21295339; http://dx.doi.org/10.1016/S0140-6736(10)62308-2
- Orrú CD, Wilham JM, Raymond LD, Kuhn F, Schroeder B, Raeber AJ, Caughey B. Prion disease blood test using immunoprecipitation and improved quaking-induced conversion. MBio 2011; 2:e00078-11; PMID:21558432; http://dx.doi.org/10.1128/mBio.00078-11
- Cardone F, Sowemimo-Coker S, Abdel-Haq H, Sbriccoli M, Graziano S, Valanzano A, Berardi VA, Galeno R, Puopolo M, Pocchiari M. Assessment of prion reduction filters in decreasing infectivity of ultracentrifuged 263K scrapie-infected brain homogenates in “spiked” human blood and red blood cells. Transfusion 2014; 54:990-5 PMID:23915063; http://dx.doi.org/10.1111/trf.12369
- Gregori L, Lambert BC, Gurgel PV, Gheorghiu L, Edwardson P, Lathrop JT, Macauley C, Carbonell RG, Burton SJ, Hammond D, et al. Reduction of transmissible spongiform encephalopathy infectivity from human red blood cells with prion protein affinity ligands. Transfusion 2006; 46:1152-61; PMID:16836562; http://dx.doi.org/10.1111/j.1537-2995.2006.00865.x
- Gregori LG, Lambert B, Rohwer R. Ability of the macopharma prion capture (P-CAPTTM) filter to remove brain PrPres in leukoreduced human RBC. Vox Sang 2006; 91s3:37-320.
- Lacroux C, Bougard D, Litaise C, Simmons H, Corbiere F, Dernis D, Tardivel R, Morel N, Simon S, Lugan S, et al. Impact of leucocyte depletion and prion reduction filters on TSE blood borne transmission. PLoS One 2012; 7:e42019; PMID:22860049; http://dx.doi.org/10.1371/journal.pone.0042019
- Lescoutra N, Sumian C, Culeux A, Durand V, Deslys JP, Comoy EE. Removal of Exogenous Prion Infectivity in Leucoreduced Red Blood Cells Unit by P-Capt (Tm) Prion Removal Filter. Vox Sang 2013; 105:197
- Clewley JP, Kelly CM, Andrews N, Vogliqi K, Mallinson G, Kaisar M, Hilton DA, Ironside JW, Edwards P, McCardle LM, et al. Prevalence of disease related prion protein in anonymous tonsil specimens in Britain: cross sectional opportunistic survey. BMJ 2009; 338:b1442; PMID:19460798; http://dx.doi.org/10.1136/bmj.b1442
- de Marco MF, Linehan J, Gill ON, Clewley JP, Brandner S. Large-scale immunohistochemical examination for lymphoreticular prion protein in tonsil specimens collected in Britain. J Pathol 2010; 222:380-7; PMID:20922767; http://dx.doi.org/10.1002/path.2767
- Hilton DA, Ghani AC, Conyers L, Edwards P, McCardle L, Ritchie D, Penney M, Hegazy D, Ironside JW. Prevalence of lymphoreticular prion protein accumulation in UK tissue samples. J Pathol 2004; 203:733-9; PMID:15221931; http://dx.doi.org/10.1002/path.1580
- HPA. Creutzfeldt-Jakob disease (CJD) biannual update. http://www.hpa.org.uk/hpr/archives/2012/hpr0612.pdf., 2012.
- Bruce ME, McConnell I, Will RG, Ironside JW. Detection of variant Creutzfeldt-Jakob disease infectivity in extraneural tissues. Lancet 2001; 358:208-9; PMID:11476840; http://dx.doi.org/10.1016/S0140-6736(01)05411-3
- Hilton DA, Ghani AC, Conyers L, Edwards P, McCardle L, Penney M, Ritchie D, Ironside JW. Accumulation of prion protein in tonsil and appendix: review of tissue samples. BMJ 2002; 325:633-4; PMID:12242174; http://dx.doi.org/10.1136/bmj.325.7365.633
- Frosh A, Smith LC, Jackson CJ, Linehan JM, Brandner S, Wadsworth JD, Collinge J. Analysis of 2000 consecutive UK tonsillectomy specimens for disease-related prion protein. Lancet 2004; 364:1260-2; PMID:15464187; http://dx.doi.org/10.1016/S0140-6736(04)17143-2
- Parliamentary Select Committee on Science and Technology. MPs launch inquiry on blood, tissue and organs screening following vCJD fears. 2013. http://www.parliament.uk/business/committees/committees-a-z/commons-select/science-and-technology-committee/news/131203-blood-tissue-and-organ-screening-tor/.
- Zeidler M, Johnstone EC, Bamber RW, Dickens CM, Fisher CJ, Francis AF, Goldbeck R, Higgo R, Johnson-Sabine EC, Lodge GJ, et al. New variant Creutzfeldt-Jakob disease: psychiatric features. Lancet 1997; 350:908-10; PMID:9314868; http://dx.doi.org/10.1016/S0140-6736(97)03148-6
- Zeidler M, Stewart GE, Barraclough CR, Bateman DE, Bates D, Burn DJ, Colchester AC, Durward W, Fletcher NA, Hawkins SA, et al. New variant Creutzfeldt-Jakob disease: neurological features and diagnostic tests. Lancet 1997; 350:903-7; PMID:9314867; http://dx.doi.org/10.1016/S0140-6736(97)07472-2
- Heath CA, Cooper SA, Murray K, Lowman A, Henry C, MacLeod MA, Stewart G, Zeidler M, McKenzie JM, Knight RS, et al. Diagnosing variant Creutzfeldt-Jakob disease: a retrospective analysis of the first 150 cases in the UK. J Neurol Neurosurg Psychiatry 2011; 82:646-51; PMID:21172857; http://dx.doi.org/10.1136/jnnp.2010.232264
- Binelli S, Agazzi P, Giaccone G, Will RG, Bugiani O, Franceschetti S, Tagliavini F. Periodic electroencephalogram complexes in a patient with variant Creutzfeldt-Jakob disease. Ann Neurol 2006; 59:423-7; PMID:16437565; http://dx.doi.org/10.1002/ana.20768
- Green AJ, Thompson EJ, Stewart GE, Zeidler M, McKenzie JM, MacLeod MA, Ironside JW, Will RG, Knight RS. Use of 14-3-3 and other brain-specific proteins in CSF in the diagnosis of variant Creutzfeldt-Jakob disease. J Neurol Neurosurg Psychiatry 2001; 70:744-8; PMID:11385008; http://dx.doi.org/10.1136/jnnp.70.6.744
- Zeidler M, Sellar RJ, Collie DA, Knight R, Stewart G, Macleod MA, Ironside JW, Cousens S, Colchester AC, Hadley DM, et al. The pulvinar sign on magnetic resonance imaging in variant Creutzfeldt-Jakob disease. Lancet 2000; 355:1412-8; PMID:10791525; http://dx.doi.org/10.1016/S0140-6736(00)02140-1
- Hill AF, Butterworth RJ, Joiner S, Jackson G, Rossor MN, Thomas DJ, Frosh A, Tolley N, Bell JE, Spencer M, et al. Investigation of variant Creutzfeldt-Jakob disease and other human prion diseases with tonsil biopsy samples. Lancet 1999; 353:183-9; PMID:9923873; http://dx.doi.org/10.1016/S0140-6736(98)12075-5
- Mead S, Wadsworth JD, Porter MC, Linehan JM, Pietkiewicz W, Jackson GS, Brandner S, Collinge J. Variant Creutzfeldt-Jakob disease with extremely low lymphoreticular deposition of prion protein. JAMA Neurol 2014; 71:340-3; PMID:24445428; http://dx.doi.org/10.1001/jamaneurol.2013.5378
- Will RG, Zeidler M, Stewart GE, Macleod MA, Ironside JW, Cousens SN, Mackenzie J, Estibeiro K, Green AJ, Knight RS. Diagnosis of new variant Creutzfeldt-Jakob disease. Ann Neurol 2000; 47:575-82; PMID:10805327; http://dx.doi.org/10.1002/1531-8249(200005)47:5<575::AID-ANA4>3.0.CO;2-W
- Heath CA, Cooper SA, Murray K, Lowman A, Henry C, MacLeod MA, Stewart GE, Zeidler M, MacKenzie JM, Ironside JW, et al. Validation of diagnostic criteria for variant Creutzfeldt-Jakob disease. Ann Neurol 2010; 67:761-70; PMID:20517937
- Haïk S, Marcon G, Mallet A, Tettamanti M, Welaratne A, Giaccone G, Azimi S, Pietrini V, Fabreguettes JR, Imperiale D, et al. Doxycycline in Creutzfeldt-Jakob disease: a phase 2, randomised, double-blind, placebo-controlled trial. Lancet Neurol 2014; 13:150-8; PMID:24411709; http://dx.doi.org/10.1016/S1474-4422(13)70307-7
- Collinge J, Gorham M, Hudson F, Kennedy A, Keogh G, Pal S, Rossor M, Rudge P, Siddique D, Spyer M, et al. Safety and efficacy of quinacrine in human prion disease (PRION-1 study): a patient-preference trial. Lancet Neurol 2009; 8:334-44; PMID:19278902; http://dx.doi.org/10.1016/S1474-4422(09)70049-3
- Doh-ura K, Ishikawa K, Murakami-Kubo I, Sasaki K, Mohri S, Race R, Iwaki T. Treatment of transmissible spongiform encephalopathy by intraventricular drug infusion in animal models. J Virol 2004; 78:4999-5006; PMID:15113880; http://dx.doi.org/10.1128/JVI.78.10.4999-5006.2004
- Parry A, Baker I, Stacey R, Wimalaratna S. Long term survival in a patient with variant Creutzfeldt-Jakob disease treated with intraventricular pentosan polysulphate. J Neurol Neurosurg Psychiatry 2007; 78:733-4; PMID:17314188; http://dx.doi.org/10.1136/jnnp.2006.104505
- Todd NV, Morrow J, Doh-ura K, Dealler S, O’Hare S, Farling P, Duddy M, Rainov NG. Cerebroventricular infusion of pentosan polysulphate in human variant Creutzfeldt-Jakob disease. J Infect 2005; 50:394-6; PMID:15907546; http://dx.doi.org/10.1016/j.jinf.2004.07.015
- Bone I, Belton L, Walker AS, Darbyshire J. Intraventricular pentosan polysulphate in human prion diseases: an observational study in the UK. Eur J Neurol 2008; 15:458-64; PMID:18355301; http://dx.doi.org/10.1111/j.1468-1331.2008.02108.x
- Newman PK, Todd NV, Scoones D, Mead S, Knight RS, Will RG, Ironside JW. Postmortem findings in a case of variant Creutzfeldt-Jakob disease treated with intraventricular pentosan polysulfate. J Neurol Neurosurg Psychiatry 2014; (Forthcoming); PMID:24554103; http://dx.doi.org/10.1136/jnnp-2013-305590
- Bishop MT, Will RG, Manson JC. Defining sporadic Creutzfeldt-Jakob disease strains and their transmission properties. Proc Natl Acad Sci U S A 2010; 107:12005-10; PMID:20547859; http://dx.doi.org/10.1073/pnas.1004688107
- Moda F, Suardi S, Di Fede G, Indaco A, Limido L, Vimercati C, Ruggerone M, Campagnani I, Langeveld J, Terruzzi A, et al. MM2-thalamic Creutzfeldt-Jakob disease: neuropathological, biochemical and transmission studies identify a distinctive prion strain. Brain Pathol 2012; 22:662-9; PMID:22288561; http://dx.doi.org/10.1111/j.1750-3639.2012.00572.x
- Barron RM, Campbell SL, King D, Bellon A, Chapman KE, Williamson RA, Manson JC. High titers of transmissible spongiform encephalopathy infectivity associated with extremely low levels of PrPSc in vivo. J Biol Chem 2007; 282:35878-86; PMID:17923484; http://dx.doi.org/10.1074/jbc.M704329200
- Piccardo P, Manson JC, King D, Ghetti B, Barron RM. Accumulation of prion protein in the brain that is not associated with transmissible disease. Proc Natl Acad Sci U S A 2007; 104:4712-7; PMID:17360589; http://dx.doi.org/10.1073/pnas.0609241104
- Head MW, Yull HM, Ritchie DL, Langeveld JP, Fletcher NA, Knight RS, Ironside JW. Variably protease-sensitive prionopathy in the UK: a retrospective review 1991-2008. Brain 2013; 136:1102-15; PMID:23550113; http://dx.doi.org/10.1093/brain/aws366
- Wilson R, Plinston C, Hunter N, Casalone C, Corona C, Tagliavini F, Suardi S, Ruggerone M, Moda F, Graziano S, et al. Chronic wasting disease and atypical forms of bovine spongiform encephalopathy and scrapie are not transmissible to mice expressing wild-type levels of human prion protein. J Gen Virol 2012; 93:1624-9; PMID:22495232; http://dx.doi.org/10.1099/vir.0.042507-0
- Kong Q, Zheng M, Casalone C, Qing L, Huang S, Chakraborty B, Wang P, Chen F, Cali I, Corona C, et al. Evaluation of the human transmission risk of an atypical bovine spongiform encephalopathy prion strain. J Virol 2008; 82:3697-701; PMID:18234793; http://dx.doi.org/10.1128/JVI.02561-07
- Béringue V, Herzog L, Reine F, Le Dur A, Casalone C, Vilotte JL, Laude H. Transmission of atypical bovine prions to mice transgenic for human prion protein. Emerg Infect Dis 2008; 14:1898-901; PMID:19046515; http://dx.doi.org/10.3201/eid1412.080941
- Sandberg MK, Al-Doujaily H, Sigurdson CJ, Glatzel M, O’Malley C, Powell C, Asante EA, Linehan JM, Brandner S, Wadsworth JD, et al. Chronic wasting disease prions are not transmissible to transgenic mice overexpressing human prion protein. J Gen Virol 2010; 91:2651-7; PMID:20610667; http://dx.doi.org/10.1099/vir.0.024380-0
- Tamgüney G, Giles K, Bouzamondo-Bernstein E, Bosque PJ, Miller MW, Safar J, DeArmond SJ, Prusiner SB. Transmission of elk and deer prions to transgenic mice. J Virol 2006; 80:9104-14; PMID:16940522; http://dx.doi.org/10.1128/JVI.00098-06
- Barria MA, Balachandran A, Morita M, Kitamoto T, Barron R, Manson J, Knight R, Ironside JW, Head MW. Molecular barriers to zoonotic transmission of prions. Emerg Infect Dis 2014; 20:88-97; PMID:24377702; http://dx.doi.org/10.3201/eid2001.130858
- Gambetti P, Dong Z, Yuan J, Xiao X, Zheng M, Alshekhlee A, Castellani R, Cohen M, Barria MA, Gonzalez-Romero D, et al. A novel human disease with abnormal prion protein sensitive to protease. Ann Neurol 2008; 63:697-708; PMID:18571782; http://dx.doi.org/10.1002/ana.21420
- Head MW, Knight R, Zeidler M, Yull H, Barlow A, Ironside JW. A case of protease sensitive prionopathy in a patient in the UK. Neuropathol Appl Neurobiol 2009; 35:628-32; PMID:19671081; http://dx.doi.org/10.1111/j.1365-2990.2009.01040.x