Abstract
HLA‑B27 plays a central role in the pathogenesis of many spondyloarthropathies and in
particular ankylosing spondylitis. The observation that the HLA‑B27 heavy chain has
a tendency to misfold has raised the possibility that associated diseases may belong in a
rapidly expanding category of protein misfolding disorders. The synthesis of the HLA‑B27 heavy
chain, assembly with b2m and the loading of peptide cargo, occurs in the endoplasmic reticulum
(ER) before transport to the cell surface. The evidence indicates that misfolding occurs in the ER
prior to b2m association and peptide optimization and is manifested in the formation of aberrant
inter‑ and intra‑chain disulfide bonds and accumulation of heavy chain bound to the chaperone
BiP. Enhanced accumulation of misfolded heavy chains during the induction of class I expression
by cytokines, can cause ER stress resulting in activation of the unfolded protein response (UPR).
Effects of UPR activation on cytokine production are beginning to emerge and may provide important
missing links between HLA‑B27 misfolding and spondyloarthritis. In this chapter we will
review what has been learned about HLA‑B27 misfolding in human cells and in the transgenic rat
model of spondyloarthritis‑like disease, considering it in the context of other protein misfolding
disorders. These studies provide a framework to support much needed translational work assessing
HLA‑B27 misfolding and UPR activation in patient‑derived material, its consequences for disease
pathogenesis and ultimately how and where to focus intervention strategies.
Introduction
Ankylosing spondylitis (AS) is a complex genetic trait with an estimated four to ten genes responsible for the majority of susceptibility.Citation1 Spondyloarthropathies (SpA) comprise several disorders that are more heterogeneous clinically and where genetic susceptibility is likely to be more complex and variable. Defining genetic loci and ultimately genes that influence susceptibility, is an area of intense investigation. Family-based linkage studies using low-density microsatellite markers have been somewhat disappointing.Citation2,Citation3 However, single nucleotide polymorphism (SNP) identification and mapping has provided a detailed framework on which to perform whole genome association studies. This approach has already provided valuable information on genes involved in susceptibility to other complex genetic diseasesCitation4 and studies on AS are now emerging.Citation5 In addition to providing markers that will be useful in identifying AS patients earlier in their disease course, it is anticipated that a more complete picture of genetic susceptibility will inform us on pathways that are important in pathogenesis and identify new therapeutic targets.
Unlike most complex genetic diseases, a single gene (HLA-B) plays a dominant role in AS. The B27 allele contributes as much as 40% of the overall genetic load and is a major factor for many other SpA.Citation6,Citation7 Although the role of HLA-B27 has been the focus of intense investigation for over 30 years, none of the postulated mechanisms has been proven or eliminated.Citation8 While it has been tacitly assumed that a single feature of HLA-B27 is responsible, it is conceivable that this is not the caseCitation9 and that the answer to the HLA-B27 conundrum will be even more complex than initially anticipated.
A detailed understanding of pathogenesis requires animal models that phenocopy the human condition and are amenable to genetic manipulation and experimentation,Citation10 combined with translational studies of human material that are confirmatory. The development of animal models has been attempted over the years through the generation of HLA-B27 transgenic mice and rats. Transgenic mouse models of SpA have been disappointing for several reasons. Initially, no spontaneous inflammatory disease was observedCitation11 and although attempts to induce disease with infection revealed some differences in susceptibility,Citation12 the SpA phenotype was not observed. Subsequent studies suggested that HLA-B27 transgenic mice developed spontaneous arthritis if you deleted the endogenous gene for (mouse) β2m.Citation13–Citation15 However, the use of a mixed genetic background may have confounded these studies making reproducibility and thus interpretation problematic.Citation16
Controlling for mixed backgrounds is very difficult and eventual genetic drift can result in loss of the phenotype. Inbred strains of animals are the genetic equivalent of a single human and thus it is not surprising that different genetic backgrounds would influence susceptibility. In humans less than 5% of HLA-B27 positive individuals develop SpA. The whole genome association studies mentioned above are being used to identify other human genes that affect susceptibility. It may be possible to exploit strain differences that are important determinants of disease in HLA-B27 transgenic rodents and use similar genetic approaches to identify the responsible genes. While the rodent genes may not be the same as those found in humans, by definition they will be involved in pathways that are important for pathogenesis of HLA-B27-associated disorders and substantial overlap with pathways identified in human genetic studies would be expected.
The production of transgenic rats expressing HLA-B27 and human β2m (B27/hβ2m) that develop spontaneous inflammation resembling SpA signified a major advance in this area.Citation17 This demonstrated that under certain conditions overexpression of HLA-B27 was sufficient to cause disease, providing the first evidence that the gene product itself was involved. The SpA-like phenotype includes gastrointestinal tract inflammation (e.g., colitis), arthritis and other inflammatory lesions in the skin and testicles. The colitis is highly penetrant, while arthritis is less frequent and depends more on the strain of rat used. Although axial inflammation can occur, it does not recapitulate the spinal inflammation and ankylosis seen in humans.Citation18 However, recently Tran et al. have reported that overexpressing additional hβ2m can alter the phenotype in transgenic rats that already overexpress HLA-B27 and hβ2m.Citation19 High copy number B27/hβ2m transgenics with additional hβ2m develop more severe arthritis and significant axial disease with no apparentchange in colitis. Interestingly, rats with low copy number B27/hβ2m that normally do not develop spontaneous disease, develop axial and peripheral arthritis without colitis, when additional hβ2m is overexpressed.
In this article we will focus on one mechanism that may be the basis for the striking relationship between HLA-B27 and spondyloarthritic diseases. We will explain the general concept and consequences of protein misfolding and then provide a detailed assessment of the special case of HLA-B27 misfolding and how it may be linked to disease through an autoinflammatory stimulus.
Usual, Unusual and Unique Features of HLA-B27 and Their Potential Role in Disease Pathogenesis
Extensive polymorphism at the HLA-B locus results in considerable sequence variation in the HLA-B-encoded heavy chain across the human population. Over 900 alleles have been reported to date, encoding over 800 different proteins (www.anthonynolan.com/HIG/). These amino acid differences affect a number of properties of class I heavy chains, including peptide binding specificity and affinity, T-cell recognition (both as a result of and independent from bound peptide), β2m binding affinity, folding and assembly efficiency and chaperone interaction (e.g., tapasin) (reviewed in ref. Citation20). There are also polymorphisms in the promoter region of HLA-B at the 5′ end of the gene, which could affect baseline expression level and inducibility.Citation21
Features of HLA-B27 that distinguish it from other alleles have provided the basis for several hypotheses concerning its contribution to disease. It is convenient to classify these ideas based on whether they invoke immunological recognition of some form of the heavy chain versus intracellular effects.Citation8 Immunological recognition by antibodiesCitation22 or autoreactive T-cellsCitation23 supposes that the form(s) of HLA-B27 being recognized are typical for HLA class I complexes.
More recently the detection of other forms of HLA-B27, such as heavy chain homodimers,Citation24 or unusual unfolded monomers,Citation25 has led to ideas about recognition by leukocyte receptors on NK cells and other leukocytes.Citation26–Citation30 In contrast, the tendency of HLA-B27 heavy chains to misfold in the intracellular compartment known as the endoplasmic reticulum (ER)Citation31,Citation32 has led to the notion that intracellular effects of HLA-B27 might be involved in disease pathogenesis. Misfolding was hypothesized to result in activation of an intracellular stress response pathway known as the unfolded protein response (UPR),Citation33 which has been shown to occur in B27/hβ2m transgenic rats.Citation34,Citation35 The consequences of HLA-B27-induced UPR activation will be discussed in detail later in this chapter. Finally, the observation that cell lines transfected with HLA-B27 but not other alleles exhibit increased bacterial survivalCitation36,Citation37 could be important for pathogenesis, particularly in reactive arthritis. Recent evidence suggests that bacterial replication is increasedCitation38 and that the p38 MAP kinase pathway may be disrupted.Citation39 This most likely represents an intracellular or at least nonantigen-presenting effect of HLA-B27.Citation40 Experiments using site-directed mutants of HLA-B27 show that the biological effect correlates with heavy chain misfolding, but is not associated with acute UPR activation and therefore the molecular mechanism remains to be defined. It will be important to determine whether the expression of heavy chains that misfold is responsible for this effect, since a related phenomenon has been observed for a mutant of surfactant protein-C that misfolds.Citation41 These authors demonstrated that adaptation to chronic ER stress was associated with modification of an NFκB-dependent pathway, reminiscent of what has been observed in HLA-B27-transfected cells.Citation42
In this chapter we will focus on HLA-B27 misfolding, considering it in the context of other proteins that misfold, the cause of misfolding and more importantly, what we have begun to learn about its consequences.
There has been a tendency to assume that only one hypothesis, or one aspect of the immunobiology of HLA-B27, will eventually be tied to its role in pathogenesis. However, this may not be correct, particularly when one considers phenotypic differences in the diseases associated with HLA-B27 such as reactive arthritis, uveitis, AS and other forms of undifferentiated SpA.
Importance of Protein Folding
The information required for a protein to fold into its native conformation is contained within its primary sequence, yet a great deal of energy is expended to ensure that this occurs efficiently and without error (reviewed in ref. Citation43). For multi-subunit proteins or those that transport cargo, the process is even more complex, with many opportunities for errors in the formation of stable, properly folded complexes. It has become increasingly apparent over the last decade that many genetic diseases result from protein misfolding, either due to inherent properties of the mutated gene product, or in some cases as a consequence of abnormalities in the cellular pathways that handle misfolded proteins.
HLA class I folding and assembly.
HLA class I complexes of heavy chain, β2m and peptide represent an example of a protein (heavy chain) that transports ‘cargo’ (β2m and peptide) to the cell surface. To perform this function and display self-peptides or pathogen-derived cargo to T-cells during an immune response, HLA class I heavy chains must fold properly, bind β2m and then load peptide prior to exiting the ER compartment (reviewed in ref. Citation44). High stability of the trimolecular complex is critical for efficient transport through the Golgi, a long half-life on the cell surface and ultimately a productive immune response. The stability of HLA class I complexes is critically dependent on early events in the folding and assembly process, including the formation of two intrachain disulfide bonds.Citation45 The α3 domain folds very rapidly and is stabilized by an intradomain disulfide between Cys-203 and Cys-259. The α1 and α2 domains fold more slowly and this is not complete until peptide is stably bound.Citation46 A second disulfide between the α1 and α2 domains (Cys-101-Cys-164) maintains the integrity of the peptide-binding grooveCitation47 as the heavy chain/β2m heterodimer interacts with tapasin, ERp57 and the transporter associated with antigen processing (TAP) to form the peptide loading complex (PLC). Although there are allelic differences in the need to interact with tapasin (and thus the PLC), in general this process facilitates the binding of high affinity peptides. For example, HLA-B27 (the B*2705 subtype) is expressed relatively efficiently in tapasin-deficient cellsCitation48 and is frequently referred to as a tapasin-independent allele. However, it interacts with tapasin when present and this affects the peptide repertoire.Citation49 It is possible that the ability of HLA-B27 to be expressed at high levels on tapasin-deficient cells may reflect its tendency to fold slowly and be retained in the ER in a peptide-receptive state without tapasin-PLC interaction. This could favor the binding and optimization of available peptides without tapasin-mediated retention.
ERp57 binds to tapasin via a disulfide (ERp57-Cys-57-Cys-95-Tapasin) and plays an important role in disulfide bond isomerization in the heavy chain during class I assembly.Citation50 Recent evidence indicates that formation of the ERp57-tapasin conjugate prevents ERp57-mediated reduction of the a1-a2 interdomain disulfide in the class I heavy chain, thus maintaining the peptide binding groove in a receptive state.Citation45 When tapasin is missing or mutated at Cys-95 and thus unable to form a complex with ERp57, the class I heavy chain a1-a2 disulfide is reduced until suitable peptide cargo can bind. Free ERp57 (or ERp57-calreticulin complexes) appears to catalyze this reduction in the absence of tapasin leading to the concept that tapasin performs its function by sequestering ERp57.
The final stages of peptide binding to HLA class I molecules includes trimming by the ER aminopeptidase associated with antigen processing (ERAP1).Citation51–Citation56 Peptides appear to be nestled into the peptide-binding groove at their C-terminus first with ERAP1-mediated N-terminal trimming to their final size of 8–11 amino acids. In humans, L-RAP or ERAP2 may also play a role in this process. In addition to peptide trimming for presentation by class I molecules, ERAP1 appears to have another role in the immune system. It was discovered independently as aminopeptidase regulator of TNF receptor (TNFR1) shedding (ARTS-1), but also regulates shedding of IL-6 and IL-1 decoy receptors.Citation57–Citation59
Consequences of Protein Misfolding
The vast majority of proteins are made in the cytosol, or cotranslationally inserted into the ER in the case of membrane bound and secreted proteins. In these two compartments, there are parallel molecular chaperone systems that assist and monitor the folding process to ensure high fidelity production of proteins that can function properly. When protein folding goes awry, due to mutations or polymorphisms that alter the amino acid sequence, or abnormalities in components of the chaperone systems, misfolding can result (reviewed in ref. Citation43). The consequences of misfolding depend on the site of synthesis of the protein, the nature and severity of the folding defect, the relative importance of the gene product and whether protein quality control (PQC) processes have intervened sufficiently. Many misfolded and even incompletely folded ER proteins can be eliminated efficiently by ER-associated degradation (ERAD) if they have remained in the ER for a sufficient time. Diseases that ensue are typically due to loss-of-function with classic examples being hemophilia (Factor VIII mutations) and hereditary emphysema (α-1-antitrypsin deficiency)Citation60 (). Gain-of-function phenotypes are more common and more varied. Misfolded proteins that accumulate within (e.g., forming aggresomes or inclusion bodies) or outside the cell (e.g., amyloid fibrils) can be toxic either to the involved cell or surrounding cells. Alpha-1-antitrypsin mutations can also cause pathology due to ER retention, aggregation and mitochondrial injury in the liver,Citation61 providing a striking example of phenotypic variation due to cell-specific differences in the handling of misfolded proteins.
The cellular response to ER protein misfolding referred to as the UPR (unfolded protein response), is part of a more global homeostatic response to ER stress.Citation60 The UPR also plays a key role in ER expansion during the differentiation of certain cell types, such as plasma cells that become highly specialized to produce and secrete large amounts of immunoglobulins.Citation62 The UPR can also contribute to the pathogenesis of certain diseases with the most clear-cut examples being situations where UPR-induced apoptosis results in the loss of important cells, such as pancreatic β-cells in the Akita mouse model of diabetesCitation63,Citation64 or neural tissue in Pelizaeus-Merzbacher disease (proteolipid protein 1 in spastic paraplegia).Citation65 Another interesting example may be idiopathic inflammatory myositis. In certain forms of the disease muscle tissue (myocytes) exhibits robust UPR activation.Citation66 This has been associated with caspase-12 activation and it has been postulated that the UPR plays a role in myositis pathogenesis by promoting apoptosis, although additional mechanisms are possible.Citation66,Citation67 Enforced class I upregulation (H-2Kb) via a tetracycline-regulated transgene driven by a muscle-specific promoter can result in an inflammatory phenotype that recapitulates much of the pathology seen with human disease.Citation68 This is interesting and may represent an example of inappropriate expression of a class I protein, perhaps with insufficient concomitant expression of peptides, β2m and/or other chaperones such as tapasin, leading to ER stress and UPR activation.
While gain-and loss-of-function classification schemes are useful, disease pathogenesis is often complex and may result from more than one consequence of protein misfolding. This is best exemplified by α-1-antitrypsin mutations that result in both types of sequelae.
HLA-B27 Misfolding
The first indication that HLA-B27 had a tendency to misfold came from mutagenesis experiments where the entire ‘B pocket’ was changed by substituting residues from the HLA-A2 allele (creating a hybrid referred to as B27.A2B).Citation69 Remarkably, this dramatically altered the folding and assembly characteristics of the heavy chain with B27. A2B behaving more like HLA-A2 and other alleles that exhibit rapid folding and assembly kinetics.Citation31 Evidence that the heavy chain was in fact misfolding, came from experiments looking at where it was being degraded. Normally, HLA class I heavy chains are internalized from the cell surface and broken down in lysosomes. However, a proportion of HLA-B27 heavy chains were found to be dislocated from the ER membrane shortly after synthesis and before becoming associated with β2m (and probably peptide) and then degraded in the cytosol by proteasomes. This ERAD pathway is used to eliminate ER-synthesized proteins that misfold and/or fail to assemble rapidly.Citation70 B27.A2B, as well as other naturally occurring HLA alleles that were examined, did not exhibit this behavior, thus tying misfolding to B pocket composition and the slow folding characteristic of HLA-B27.Citation32 ERAD of HLA-B27, but not the other expressed alleles, was also detected in EBV-transformed human B-cells indicating that it occurs when there is only a single copy of the HLA-B27 gene and is not merely a consequence of overexpression.Citation31
Interestingly, the B pocket was also found to have an unexpected dramatic effect on peptide binding affinity, in addition to its predicted effect on peptide binding specificity.Citation31 Since this pocket binds the side chain of the second amino acid in the peptide (P2), the specificity conferred by the HLA-A2-like B pocket was almost identical to what is found in peptides that bind to HLA-A2 (Leu/Met), rather than the Arg P2 specificity of HLA-B27. However, what was surprising was that HLA-B27 required a 30-fold higher concentration of peptide (on average) to achieve the same half-maximal binding as B27.A2B. This suggests that the folding abnormality exhibited by HLA-B27 may be related to peptide binding. In other words, this allele might require more peptide to achieve release from the PLC and exit from the ER. It would follow that in situations where the supply of peptides into the ER is restricted and/or the synthesis of heavy chains is increased, HLA-B27 folding might be disproportionately adversely affected in comparison to other alleles.
Further exploration of events occurring in the ER for HLA-B27 revealed that the heavy chain has a tendency to form disulfide-linked complexes with itself (and possibly other proteins; unpublished observations) and exhibit prolonged association with the ER chaperone BiP (Grp78/Hspa5).Citation32,Citation71,Citation72 These features also map to the B pocket and are not exhibited by B27.A2B or other naturally occurring alleles examined to date. Further mutagenesis experiments have defined two B pocket residues that are key for HLA-B27 misfolding; Glu-45 and Cys-67 (reviewed in ref. Citation73). The single substitution of Met for Glu at position 45 restores rapid folding to the HLA-B27 molecule and eliminates the formation of disulfide-linked complexes and prolonged BiP binding (misfolding) even in the presence of Cys-67. Furthermore, the single substitution of Ala for Cys at position 67 also prevents misfolding, even when Glu-45 is intact. These observations suggested a model where two features of the HLA-B27 heavy chain might be required for misfolding to be prominent; slow folding and the ability to form aberrant disulfide-linked dimers via Cys-67.Citation73
While Glu-45 and Cys-67 are not unique to HLA-B27, they are very uncommon among other alleles. In addition, there is a Lys residue at position 70 that has been reported to increase the reactivity of the sulfhydryl group on Cys-67,Citation74 although it has not been studied in the context of misfolding. These three residues (Glu-45, Cys-67 and Lys-70) are virtually unique to HLA-B27,Citation75 (www.anthonynolan.com/HIG/) and thus would support the idea that misfolding is extremely uncommon if not unique to this allele.
Additional support for this model comes from the observation that an imposed reduction in folding rate caused by incubating cells at reduced temperature also exacerbates dimer formation and BiP binding to heavy chains.Citation72 In this study, evidence was provided that Cys-164, in addition to Cys-67, was involved in dimer formation. This observation has several possible implications since Cys-164 is involved in the intrachain disulfide bridge between the α1 and α2 domains of the class I heavy chain (Cys-101-Cys-164), which normally forms quite rapidly after heavy chain synthesis and is important for maintaining the integrity of the peptide-binding groove (see above).
The involvement of Cys-164 residue in oxidative dimerization of HLA-B27 heavy chains is potentially important as it suggests two possible scenarios related to HLA-B27 misfolding. First, if the Cys-101-Cys-164 disulfide bridge forms quickly in HLA-B27 as in other alleles, then it must not be completely protected from reduction/isomerization if it is eventually involved in dimerization, since the latter process requires it to reform a disulfide with another HLA-B27 heavy chain. Since protection of the Cys-101-Cys-164 disulfide from reduction is a key function of tapasin-ERp57,Citation45 the formation of dimers via Cys-164 could reflect HLA-B27 not interacting efficiently with this complex in the ER. Alternatively, it may be that the a1-a2 domain disulfide does not form normally in HLA-B27, making Cys-164 available to enter into an interchain disulfide linkage. Additional studies are needed to fully delineate the earliest events in HLA-B27 folding that lead to misfolding and its cellular consequences.
Evaluating the Role of HLA-B27 in Disease
A major advance toward understanding the role of HLA-B27 in SpA was made in the 1990s when Taurog and colleagues first produced transgenic rats overexpressing HLA-B27 and human β2m (hβ2m) (B27/hβ2m).Citation17 High copy number B27 transgenic rats were found to develop a ‘spontaneous’ inflammatory disease that includes gastrointestinal inflammation (colitis), arthritis, alopecia with psoriasis-like skin lesions, dystrophic nails and testicular inflammation.Citation18 These phenotypic features are only partially penetrant and are variable in frequency with the exception of colitis, which occurs in 100% of transgenics. The arthritis is predominantly peripheral, although rats overexpressing additional hβ2m were shown recently to develop more severe arthritis with axial involvementCitation19 (discussed below). While transgenic rats do not provide a precise phenocopy of human disease, B27/hβ2m transgenics with and without extra hβ2m provide reproducible animal models that can be used to investigate pathogenic mechanisms that are likely to have relevance to human disease. Unfortunately rats are not as amenable to experimental manipulation as mice.
For example, targeted gene deletion is not currently possible due to the lack of embryonic rat stem cells. Production of transgenics is more expensive and labor intensive, ex vivo transduction of bone marrow cells with retroviruses is not readily achievable and many antibodies useful to visualize and/or block the function of mouse proteins are not available for rats. Nevertheless, a great deal has been learned about the cellular requirements for disease in high copy B27/hβ2m transgenic rats (reviewed in ref. Citation18). HLA-B27 must be expressed in the bone marrow compartment for the colitis/peripheral arthritis phenotype to occur and ubiquitous expression is not necessary.Citation76 In addition, the spontaneous inflammatory disease appears to be mediated by CD4 rather than CD8 T-cells.Citation77,Citation78 The presence of gastrointestinal flora is also required, yet normal flora is sufficient to trigger inflammation, especially bacteroides spp.Citation79,Citation80 These findings have provided strong evidence against a role for arthritiogenic (or ‘colitogenic’) peptides playing a central role in pathogenesis, but rather suggest that HLA-B27-expressing cells arising from the bone marrow are either targeted by CD4 T-cells or alternatively serve as a stimulus for these cells to become pathogenic.
Is HLA-B27 recognized by CD4 T-cells?
Reports that CD4 T-cells can recognize normal and abnormal forms of HLA-B27 have emerged,Citation81 raising the question of whether this might explain the importance of these cells for SpA-like disease in B27/hβ2m transgenic rats and also be involved in human disease. For human studies, CD4 T-cells were raised by stimulation with T2 cells transfected with HLA-B27.Citation81 T2 cells are missing a large region of the major histocompatibility complex (MHC) including TAP1 and TAP2 genes and thus are unable to transport peptides into the ER from the cytosol. They have been reported to express HLA-B27 homodimers,Citation24 although this was not observed in other studies.Citation32,Citation72
Evidence supporting the idea that CD4 T-cells could recognize HLA-B27 came from experiments using a monoclonal antibody (ME1) that recognizes HLA-B27 and could block recognition. When cells with an intact antigen presentation pathway were used, including patient-derived B-cells, HLA-B27 was poorly recognized. In a follow-up study CD4 T-cells from two more AS patients were raised using similar methods.Citation82 These T-cells failed to recognize HLA-B27 on T2 cells, but instead appeared to be reacting to other HLA class I alleles expressed at low levels on these cells, perhaps presenting peptides derived from degraded HLA-B27 heavy chains. In separate studies, double transgenic mice expressing HLA-B27 and a human T-cell receptor (TCR) that recognizes the HLA-B27-restricted NP383-391 flu peptide, developed CD4 as well as CD8 T-cells capable of recognizing this peptide presented by HLA-B27.Citation83 If CD4 T-cells that can recognize HLA-B27 develop in rats and humans, this could have implications for disease. However, these transgenic mice represent an unusual situation where there is forced expression of the same TCR on every T-cell regardless of the costimulatory CD4/8 molecule and thus the question of whether this might occur with TCRs directed against other alleles needs to be addressed. The possibility that CD4 T-cells capable of recognizing HLA-B27 exist in transgenic rats has not, to our knowledge, been examined.
What are the consequences of HLA-B27 misfolding?
The observation that HLA-B27 had a propensity to misfold, as defined by the formation of disulfide-linked heavy chains and stable BiP binding, was confirmed and extended in B27/hβ2m transgenic rats.Citation71 Using several transgenic lines with variable transgene copy number and phenotype, Tran et al. demonstrated a quantitative correlation between the biochemical features of HLA-B27 misfolding in splenocytes and the development of SpA-like disease (colitis and arthritis). This correlation was further supported by the absence of disease in HLA-B7 (B7/hβ2m) transgenic rats, consistent with the evidence that this allele does not misfold, even when overexpressed.Citation32
The unfolded protein response.
One of the consequences of protein misfolding in the ER can be activation of the UPR (reviewed in ref. Citation84). Some of the earliest cellular events that mark this response are phosphorylation and activation of PERK (PKR-like ER kinase) and IRE1 (inositol requiring-1) and proteolytic cleavage of ATF6 (activating transcription factor-6). Immediate downstream events include IRE1-mediated splicing of the mRNA encoding XBP-1 (X-box binding protein-1), PERK-mediated phosphorylation of eIF2α (eukaryotic initiation factor 2 α) and increased transcription of UPR target genes (e.g., BiP, CHOP and many others). The transcriptional response is mediated by activated (cleaved) ATF6, ATF4 (produced in response to eIF2a phosphorylation) and the gene product translated from the spliced XBP-1 mRNA (XBP-1s), all of which are active transcription factors.
Several reagents used to measure proximal UPR activation (e.g., antibodies to ATF6 and phosphorylated forms of PERK and IRE1) are not available for rats. Furthermore, since the response is transient, it is more convenient to assess induction of mRNAs encoding BiP and CHOP and splicing of XBP-1 transcripts (XBP-1s). Using these markers, we found that spleen and thymus cells isolated from B27/hβ2m transgenic rats (F344 33.3 line) exhibited little or no evidence of UPR activation.Citation34 Similarly, bone marrow (BM)-derived macrophages from premorbid rats showed minimal differences in BiP mRNA (50% or 1.5-fold increase), whereas when BM macrophages were prepared from animals with disease, a robust UPR was observed (5-fold increase in BiP mRNA and up to a 10-fold increase in CHOP). Microarray analyses revealed the UPR to be accompanied by an interferon (IFN) signature raising the question of whether IFN exposure is causing UPR activation via HLA-B27 upregulation. The converse was also possible: the UPR might cause IFN upregulation. It was conceivable that both events were occurring simultaneously.
IFN regulation of HLA-B27 expression and UPR activation.
Subsequent studies have revealed a dual role for IFNs in UPR activation in BM macrophages from B27/hβ2m transgenic rats. First, BM macrophages expressing HLA-B27 that exhibit no UPR at ‘baseline’ (i.e., without stimulation) will activate the UPR in response to IFN-γ treatment.Citation34,Citation35
This is temporally related to heavy chain upregulation and accompanied by a striking increase in the accumulation of BiP-bound heavy chains and disulfide-linked heavy chain complexes.Citation35 In contrast, IFN-γ does not activate the UPR in cells from nontransgenic (wild type) or B7/hβ2m transgenic animals. (It should be noted that there is low-level BiP induction and XBP-1 splicing (<2-fold increase) with IFN-γ treatment of macrophages from these animals, but the response in B27/hβ2m transgenics is substantially higher.Citation34 IFN-γ has been reported to cause ER stress in oligodendrocytes, but this response was also quantitatively small (∼2-fold BiP induction) and required prolonged (48 h) stimulation.Citation85 This is not surprising given that cytokines and other exogenous stimuli can upregulate hundreds of proteins, including membrane bound and secreted proteins that are produced in the ER. This low level UPR is likely part of the normal physiologic response that enables cells to handle the increased load. It is worth emphasizing that exacerbated HLA-B27 misfolding and UPR activation occur in the face of IFN-γ-mediated upregulation of multiple components of the class I assembly pathway including TAP1/2, tapasin, proteasome subunits LMP2 and LMP7, ERAP1 and β2m. This implies that HLA-B27 misfolding and ER stress occur despite an increased source of cargo (β2m and peptide) as well as equipment necessary to load the cargo. This seems paradoxical and could indicate that one or more of these components exacerbate HLA-B27 misfolding, although an alternative explanation is that they may merely be insufficient to prevent misfolding.
When splenocytes are treated with IFN-γ, we see only low-level upregulation of HLA-B27 and minimal UPR activation. Examination of inflamed colon tissue reveals evidence for UPR activation, although the magnitude of increases in BiP and CHOP transcripts are smaller (<3-fold) than observed in isolated cells such as BM macrophages (reviewed in ref. Citation34). Together, these data indicate that UPR activation occurs in cells and inflamed tissues from B27/hβ2m transgenic rats, is specific for HLA-B27 and is temporally related to and strongly correlated with HLA-B27 misfolding. Macrophages are particularly affected by HLA-B27 misfolding in terms of UPR activation, while splenocytes, whole spleen and whole thymus tissue are not.Citation35 These results are consistent with HLA-B27 upregulation being a key component of robust UPR activation.
In preliminary studies we have observed UPR activation in BM-derived dendritic cells (DCs) from B27/hβ2m transgenic rats treated with IFN-γ, but it does not appear to be as robust as in macrophages. However, since additional stimuli can contribute to class I upregulation and we have not exhaustively examined other cell types, our understanding of the extent of UPR activation in these rats remains incomplete.
IFN induction by the UPR.
The second part of the IFN story, is the question of whether IFN expression is upregulated by the UPR. We found low-level induction of the Type I IFN, IFN-β, in BM macrophages undergoing a UPR, either due to HLA-B27 upregulation or in cells treated with pharmacologic agents (tunicamycin or thapsigargin) that cause ER stress,Citation86 consistent with a previous report of low-level induction in tunicamycin-treated fibroblasts.Citation87 IFN-β has well-recognized autocrine effects at low concentrations,Citation88–Citation90 and thus UPR-induced IFN-β may have immunological consequences including a pro-survival effect on macrophages.Citation91 However, perhaps more important is the response observed when macrophages undergoing a UPR are exposed to ligands for pattern recognition receptors (e.g., Toll-like receptors or TLRs). TLR4 and TLR3 agonists such as LPS and dsRNA, that upregulate IFN-β via the TRIF (Toll-like receptor/IL-1 receptor related adaptor protein inducing IFN-β)-dependent pathway, cause robust synergistic IFN-β production in cells exhibiting ER stress. The synergistic response appears to require XBP-1s, but not PERK or ATF6 activation. These results suggest a fundamental relationship between ER stress and innate immune signaling with implications beyond HLA-B27 and disease, as well as a novel function of XBP-1 in the convergence of these important signaling pathways.
The UPR and cytokine production.
Links between the UPR and cytokine induction have been reported in the literature. IL-6 production from plasma cells after activation by LPS or CD40 ligation is influenced by XBP-1, although this effect is considerably delayed and may be secondary to other changes.Citation62 Macrophages loaded with cholesterol exhibit UPR activation and increased production of TNF-α and IL-6, effects that appear to be secondary to NFκB, JNK1/2, p38 and/or Erk1/2 activation.Citation92 Using a microarray-based screening approach, we identified IL-23p19 (the unique subunit of the active IL-23 cytokine), as being synergistically induced by LPS-treatment of cells with an active UPR (reviewed in ref. Citation86). We have found IL-23p19 upregulation in inflamed tissue and myeloid cells derived from the tissue, in B27/hβ2m transgenic rats. Il-23p19 is upregulated in a temporal and spatial manner that is consistent with it being involved in the development of colon inflammation. In addition, there is robust upregulation of IL-17 in the inflamed colon that localizes to CD4 T-cells in the lamina propria and draining mesenteric lymph nodes (reviewed in ref. Citation93). These findings are of interest in the context of several recent developments in our understanding of T-cell biology, as well as new evidence for genes involved in susceptibility to AS.Citation5
Th1, Th2 and Th17.
Upon antigenic stimulation, naïve CD4 T-cells differentiate into T helper (Th) cells with specialized cytokine production profiles and effector functions. The Th1/Th2 paradigm established over 20 years ago was that Th1 cells produce large quantities of IFN-γ and are essential for clearing intracellular pathogens, while Th2 cells produce IL-4, 5 and 13 and are important for clearance of extracellular organisms and robust humoral immunity.Citation94,Citation95 Key cytokines that drive these two pathways are IL-12 (IL-12p70) and IL-4. IL-12 induces Th1 differentiation through STAT4 activation in T-cells and IL-4 promotes Th2 development through STAT6 and GATA-3 activation, promoting more IL-4 production.Citation96 IFN-γ from an initial innate immune response (e.g., activated NK cells) is also important for activating the T-bet transcription factor through STAT1 signalling, which in turn activates Th1-specific genes.
Recently, a third subset of effector CD4 T-cells characterized by IL-17 production (‘Th17’) has been discovered.Citation97–Citation99 Th17 cells may have evolved as another arm of the adaptive immune response for enhanced protection against extracellular bacteria (i.e., Klebsiella pneumoniae), protozoa and fungi (e.g., Pneumocystis carinii) by recruiting neutrophils. However, additional roles for Th17 in immune defense are possible. What has become very clear, is that Th17 cells play a crucial role in chronic inflammation in animal models of human autoimmune/autoinflammatory diseases such as RA, MS,Citation100,Citation101 IBDCitation102,Citation103 and psoriasisCitation104 and there is growing evidence that IL-17 is a crucial pro-inflammatory cytokine in the human disease counterparts. In addition to IL-17, Th17 cells can produce TNF-α and IL-6.Citation100,Citation102 IL-17 can act on several cell types including macrophages, fibroblasts, endothelial cells and epithelial cells, to upregulate TNF-α, IL-6, IL-1, as well as several chemokines and metalloproteases (including MMP-3 which has been shown to be a good biomarker for AS).Citation105–Citation107 Thus, downstream effects of IL-17 are diverse and highly proinflammatory.
Several cytokines play key roles in Th17 development and the balance between Th17 and regulatory T-cells (Treg) in mice. For example, the combination of TGF-β and IL-6 drives naïve CD4 T-cells to become Th17-committedCitation108,Citation109 through induction of the retinoic acid orphan receptor (RORγt) in naïve T-cells, which then leads to upregulation of the IL-23 receptor (IL-23R).Citation110 IL-23 can then act on Th17-competent cells stimulating robust and prolonged IL-17 upregulationCitation111,Citation112 (and reviewed in ref. Citation113). In addition, TCR stimulation by MHC class II-restricted antigens can induce IL-17 production without IL-23.
In mice it appears that CD4 T-cells producing IFN-γ (Th1) and IL-17 (Th17) are distinct populations, while in humans CD4 T-cells producing IFN-γ and IL-17 (Th1/Th17) have been documented in the gut of humans with Crohn's disease.Citation114 In addition, the factors that regulate Th17 development in humans appear to be different from mice with IL-23 and IL-1β playing a more important role than IL-6.Citation115 In addition to the predominant form of IL-17 (IL-17A or CTLA-8) produced by CD4 T-cells, there is an extended family with five additional IL-17 molecules whose cellular source and regulation need to be further defined.Citation107 Other cells that have been reported to produce IL-17 include CD8 and gamma/delta T-cells, neutrophils and even macrophages and lymphocytes at sites of infection.
HLA-B27 misfolding, the UPR and the IL-23/IL17 axis: refining the hypothesis.
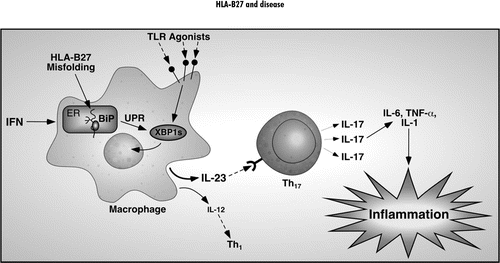
Preliminary results linking HLA-B27 misfolding and the UPR to enhanced IL-23 induction in macrophages in response to TLR agonists, together with evidence for activation of the Th17 axis in transgenic rats, suggests a novel paradigm for the development of HLA-B27-associated colitis (). In the gastrointestinal tract, a low level immune response to bacterial colonization could result in increased expression of IFNs (Type I and/or Type II), perhaps via innate immune stimuli (IFN-β) and/or NK cell activation (IFN-γ). This would result in upregulation of class I expression and, in cells expressing HLA-B27, activation of the UPR. Macrophages would then become sensitized to TLR agonists such as LPS and other bacterial products, polarizing them toward increased production of IFN-β and IL-23 and possibly more IL-6. IL-23 would then drive IL-17 production from CD4 T-cells that have become committed to the Th17 lineage. While IL-6 and TGF-β have been shown to be important for the development of Th17 T-cells,Citation99 there is evidence that cells with the capacity to produce IL-17 are normally present in the colon.Citation102,Citation103 Thus, in this unique mucosal environment, increased IL-23 expression could be a sufficient stimulus for chronicgastrointestinal inflammation. This is supported by the observation that IL-23p19 transgenic mice develop widespread inflammation without any other additional stimulus.Citation116 In the HLA-B27 transgenic rats, increased IFN-β expression might serve to promote HLA-B27 upregulation and also activate NK cells.Citation117 It is also possible that unusual forms of HLA-B27 expressed on the cell surface might engage leukocyte receptors and serve as an activating stimulus for NK cells.Citation30
It is interesting to note that IFN-γ can inhibit the Th17 axis.Citation97,Citation98 In the model we propose for the development of colitis, IFNs would play an important role in promoting IL-23 production via class I upregulation and subsequent UPR activation, but could conceivably inhibit the IL-23/IL-17 axis through effects on Th17 development. We and others, have documented IFN-γ overexpression in the inflamed colon,Citation18,Citation34 but since CD4 T-cells with the capacity to produce IL-17 may already exist in this location, IFN-γ may have little effect on their development. Furthermore, we do not know the relative importance of Type I vs Type II IFNs in HLA-B27 upregulation in rats in vivo, nor whether Type I IFNs have the same inhibitory effect on Th17 development, although this might be expected given the overlap in effects of Type I and Type II IFNs. These and other questions, including the relative importance of the Th1 axis in transgenic rats, need to be further addressed.
HLA-B27 Subtypes and Spondyloarthritis
There is heterogeneity within the HLA-B27 group of alleles referred to as subtypes (www.anthonynolan.com/HIG/). The numerical classification for subtypes is to designate them with an asterisk preceding the number (e.g., B*2701, B*2702, etc.,). More than 30 subtypes have been reported for HLA-B27 and since most occur infrequently, little is known about their association with AS or SpA. While most of the relatively common subtypes (e.g., B*2705, B*2702, B*2704) have been associated with disease, for some time B*2706 and B*2709 have been thought to be exceptions. Since hypotheses explaining how HLA-B27 might cause disease have been driven by our understanding of how it differs from other HLA-B alleles, it should be possible to refine our ideas based on properties of subtypes differentially associated with disease. However, the caveat with this approach is that incomplete or incorrect information about disease associations may lead to incorrect conclusions. There are now new data suggesting that B*2709 may be associated with disease, or at the very least the situation is more complex than previously thought.Citation118 Patients with B*2709 who developed SpA were reported several years ago,Citation119,Citation120 and now there are reports of this subtype in AS patients.Citation121,Citation122 A recent examination of the existing data suggests that B*2709 occurs in these individuals at a greater frequency than by chance alone,Citation118 thus supporting the idea that this subtype may indeed be associated with disease. The occurrence of B*2709 on a distinct haplotype from B*2705 in the same population,Citation123 along with genetic evidence that additional MHC-encoded genes influence susceptibility,Citation3,Citation124 raises the possibility that the offending alleles are not present on the B*2709 haplotype.Citation123 It is well known that most individuals with HLA-B27 (and B*2705 by inference) do not develop AS/SpA and HLA-B27-positive family members of patients with AS are at much higher risk for disease than the general HLA-B27-positive population. One recently proposed hypothesis is that B*2709 arose by a single mutation from B*2705 on a low-risk haplotype and that it may be the low-risk haplotype that is more important for disease predisposition than the immunobiological differences between the B*2705 and B*2709 proteins.Citation118 Additional genetic differences between populations with B*2709 and B*2705 might also contribute.
The case for a lack of association between B*2706 and disease is more compelling, in part because it is present in a much larger and probably more genetically diverse population.Citation125 However, patients with AS and this subtype have also been reportedCitation126 with two additional cases described recently.Citation127
Subtype associations (or lack thereof ) need to be extended to larger populations and investigated for the possible existence of MHC haplotypes such as those uncovered in Sardinia.Citation123
Another pitfall of using the genetic association data to drive hypotheses about disease causation is that subtyping of HLA-B27 has traditionally focused on coding sequence variation, with little attention to the promoter region of the gene. Promoter polymorphisms, which are known to exist,Citation21 could have consequences for baseline and inducible HLA-B27 subtype expression.
Overexpression of Additional hβ2m: The New Model of SpA
The phenotype exhibited by high copy B27/hβ2m transgenic rats, where colitis and peripheral arthritis predominate, does not include an important component of AS—axial inflammation and ankylosis. Recently Tran et al. found that overexpressing more hβ2m by introducing an additional 35 copies of the hβ2m transgene altered the phenotype of high copy B27/hβ2m transgenic rats (55 copies of HLA-B27 and 66 copies of hβ2m).Citation19 Rats with 55 copies of HLA-B27 and 101 copies of hβ2m had more frequent and more severe arthritis involving the axial skeleton, while colitis was not affected. In addition, the extra 35 copies of hβ2m were able to induce arthritis in intermediate copy B27/hβ2m transgenic rats (20 copies of HLA-B27 and 15 copies of hβ2m) that normally remain free of any spontaneous disease. Thus, spondylitis was induced by additional hβ2m even in the absence of colitis. These observations are potentially important as they provide a model system that may be relevant to the pathogenesis of axial inflammation.
The mechanism by which additional hβ2m modifies the phenotype of B27/hβ2m transgenic rats is not clear. Based on observations that the additional hβ2m increased the folding kinetics of HLA-B27, reduced the formation of aberrant disulfide linked heavy chain complexes and resulted in a reduction of BiP mRNA expression (∼25–30%) in splenocytes, the authors concluded that while HLA-B27 misfolding was still associated with intestinal inflammation, it was not critical to the development of HLA-B27-associated arthropathy. However, this conclusion is premature, since UPR activation was not examined after upregulation of HLA-B27, which we have shown is critical for this response.Citation34,Citation35 In addition, since there is some cell type specificity to HLA-B27-induced UPR activation, it will be important to examine cells that are likely to be relevant to disease pathogenesis. Preliminary experiments suggest that while the additional hβ2m reduces the magnitude of UPR activation when HLA-B27 is upregulated, it does not eliminate it (unpublished observations) and thus the role of HLA-B27 misfolding in the spondyloarthritis phenotype will require further investigation.
It is also important to consider that UPR activation might be a ‘double-edged’ sword in the pathogenesis of inflammatory disease. Its consequences could depend on the magnitude of the response. For example, it is well known that a strong and unresolved UPR can lead to apoptosis.
If UPR activation in macrophages drives an inflammatory process due to abnormal cytokine production, one could envision downstream effects being different if the cells causing the problem are destined to undergo UPR-induced apoptosis. The consequences of inappropriate in vivo UPR activation in the immune system are relatively unexplored and it is also likely that we do not yet appreciate precisely what needs to be examined. Our ability to approach these questions would be aided greatly by the development of a mouse model, where many more tools are available to address these complex issues.
Conclusions
Recent advances in deciphering genetic susceptibility to AS point toward the IL-23 receptor (IL23R) gene.Citation5 This gene encodes a protein that combines with another subunit IL-12Rβ1 to form the active IL-23 receptor expressed on developing Th17 T-cells,Citation113 making them responsive to IL-23. IL23R polymorphisms have also been implicated in susceptibility to Crohn's disease and psoriasis, other diseases that have phenotypic overlaps with spondyloarthritis.Citation128,Citation129 Preliminary data indicating that HLA-B27 misfolding may be a stimulus for activating the IL-23/IL-17 axis, suggests a novel mechanism that may explain, at least in part, the role of HLA-B27 in colitis in transgenic rats. The striking convergence of the human genetic data and results from HLA-B27 transgenic rats provides a compelling argument that this axis needs to be further examined in SpA and AS.
Figures and Tables
Figure 1 Consequences of protein misfolding. Proper folding of newly synthesized proteins is critical for normal function. Protein misfolding has been linked to a number of diseases that can be broadly categorized as loss-of-function or gain-of-function. Loss-of-function phenotypes result from destruction of partially folded or misfolded proteins by elaborate quality control processes. Gain-of-function phenotypes can result from toxicity if the gene product accumulates and/or activation of cellular stress response pathways such as the UPR. HLA-B27 misfolding is hypothesized to result in gain-of-function abnormalities through sensitization of immune response cells such as macrophages to other exogenous stimuli as reviewed in this chapter.
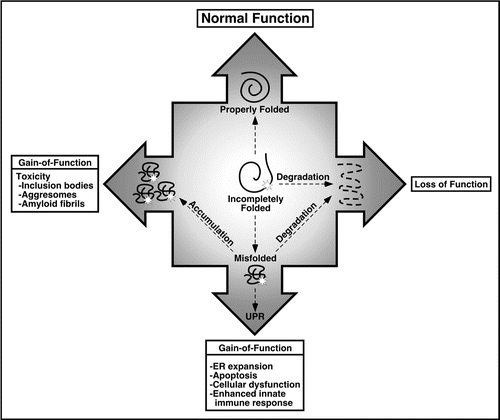
Figure 2 Proposed paradigm linking HLA-B27 misfolding to innate immune activation. The tendency of HLA-B27 to misfold and activate the UPR when upregulated sensitizes cells to certain pathogen-associated molecular patterns and possibly damage-associated molecular patterns, many of which signal through pattern recognition receptors such as the Toll-like receptors (TLR Agonists). Enhanced upregulation of IL-23 promotes IL-17 production from CD4 T-cells of the Th17 lineage. Th17 cells can produce TNFα and IL-6 and IL-17 is also a potent pro-inflammatory cytokine that acts on many tissue cell types and further induces TNFα, IL-6 and IL-1 as well as chemokines and metalloproteinases. IL-17 is hypothesized to be a key pro-inflammatory cytokine in the immunopathology that develops in the colon of HLA-B27 transgenic rats.
Note
This manuscript has been previously published: Colbert RA, Delay ML, Layh-Schmitt G, Sowders DP. HLA-B27 misfolding and spondyloarthropathies. In: Molecular Mechanisms of Spondyloarathropathies. López-Larrea, C and Díaz-Peña, R ed. Austin and New York: Landes Bioscience and Springer Science and Business Media, 2009 In Press.
References
- Brown MA, Laval SH, Brophy S, Calin A. Recurrence risk modelling of the genetic susceptibility to ankylosing spondylitis. Ann Rheum Dis 2000; 59:883 - 886
- Laval SH, Timms A, Edwards S, Bradbury L, Brophy S, Milicic A, et al. Whole-genome screening in ankylosing spondylitis: Evidence of NonMHC genetic-susceptibility loci. Am J Hum Genet 2001; 68:918 - 926
- Zhang G, Luo J, Bruckel J, Weisman MA, Schumacher HR, Khan MA, et al. Genetic studies in familial ankylosing spondylitis susceptibility. Arthritis Rheum 2004; 50:2246 - 2254
- Sladek R, Rocheleau G, Rung J, Dina C, Shen L, Serre D, et al. A genome-wide association study identifies novel risk loci for type 2 diabetes. Nature 2007; 445:881 - 885
- Burton PR, Clayton DG, Cardon LR, Craddock N, Deloukas P, Duncanson A, et al. Association scan of 14,500 nonsynonymous SNPs in four diseases identifies autoimmunity variants. Nat Genet 2007; 39:1329 - 1337
- Reveille JD. Major histocompatibility genes and ankylosing spondylitis. Best Pract Res Clin Rheumatol 2006; 20:601 - 609
- Brown MA. Non-major-histocompatibility-complex genetics of ankylosing spondylitis. Best Pract Res Clin Rheumatol 2006; 20:611 - 621
- Smith JA, Marker-Hermann E, Colbert RA. Pathogenesis of ankylosing spondylitis: Current concepts. Best Pract Res Clin Rheumatol 2006; 20:571 - 591
- Lopez de Castro JA. HLA-B27 and the pathogenesis of spondyloarthropathies. Immunol Lett 2007; 108:27 - 33
- Breban M, Hacquard-Bouder C, Falgarone G. Animal models of HLA-B27-associated diseases. Curr Mol Med 2004; 4:31 - 40
- Kievits F, Ivanyi P, Krimpenfort P, Berns A, Ploegh HL. HLA-restricted recognition of viral antigens in HLA transgenic mice. Nature 1987; 329:447 - 449
- Nickerson CL, Luthra HS, Savarirayan S, David CS. Susceptibility of HLA-B27 transgenic mice to yersinia enterocolitica infection. Hum Immunol 1990; 28:382 - 396
- Khare SD, Luthra HS, David CS. Spontaneous inflammatory arthritis in HLA-B27 transgenic mice lacking β2-microglobulin: a model of human spondyloarthropathies. J Exp Med 1995; 182:1153 - 1158
- Khare SD, Hansen J, Luthra HS, David CS. HLA-B27 heavy chains contribute to spontaneous inflammatory disease in B27/human β2-microglobulin (b2m) double transgenic mice with disrupted mouse b2m. J Clin Invest 1997; 98:2746 - 2755
- Khare SD, Bull MJ, Hanson J, Luthra HS, David CS. Spontaneous inflammatory disease in HLA-B27 transgenic mice is independent of MHC class II molecules: a direct role for B27 heavy chains and not B27-derived peptides. J Immunol 1998; 160:101 - 106
- Kingsbury DJ, Mear JP, Witte DP, Taurog JD, Roopenian DC, Colbert RA. Development of spontaneous arthritis in β2-microglobulin-deficient mice without expression of HLA-B27: association with deficiency of endogenous major histocompatibility complex class I expression. Arthritis Rheum 2000; 43:2290 - 2296
- Hammer RE, Maika SD, Richardson JA, Tang JP, Taurog JD. Spontaneous inflammatory disease in transgenic rats expressing HLA-B27 and human b2-m: an animal model of HLA-B27-associated human disorders. Cell 1990; 63:1099 - 1112
- Taurog JD, Maika SD, Satumtira N, Dorris ML, McLean IL, Yanagisawa H, et al. Inflammatory disease in HLA-B27 transgenic rats. Immunol Rev 1999; 169:209 - 223
- Tran TM, Dorris ML, Satumtira N, Richardson JA, Hammer RE, Shang J, et al. Additional human beta(2)-microglobulin curbs HLA-B27 misfolding and promotes arthritis and spondylitis without colitis in male HLA-B27-transgenic rats. Arthritis Rheum 2006; 54:1317 - 1327
- Hildebrand WH, Turnquist HR, Prilliman KR, Hickman HD, Schenk EL, McIlhaney MM, et al. HLA class I polymorphism has a dual impact on ligand binding and chaperone interaction. Hum Immunol 2002; 63:248 - 255
- Yao Z, Volgger A, Scholz S, Albert ED. Sequence polymorphism in the HLA-B promoter region. Immunogenetics 1995; 41:343 - 353
- Yu DY, Choo SY, Schaack T. Molecular mimicry in HLA-B27-related arthritis. Ann Int Med 1989; 111:581 - 591
- Benjamin RJ, Parham P. Guilt by association: HLA-B27 and ankylosing spondylitis. Immunol Today 1990; 11:137 - 142
- Allen RL, O'Callaghan CA, McMichael AJ, Bowness P. Cutting edge: HLA-B27 can form a novel beta2-microglobulin-free heavy chain homodimer structure. J Immunol 1999; 162:5045 - 5048
- Malik P, Klimovitsky P, Deng LW, Boyson JE, Strominger JL. Uniquely conformed peptide-containing beta2-microglobulin-free heavy chains of HLA-B2705 on the cell surface. J Immunol 2002; 169:4379 - 4387
- Edwards JCW, Bowness P, Archer JR. Jekyll and Hyde: The transformation of HLA-B27. Immunol Today 2000; 21:256 - 260
- Kollnberger S, Bird L, Sun MY, Retiere C, Braud VM, McMichael A, et al. Cell surface expression and immune receptor recogntion of HLA-B27 homodimers. Arth Rheum 2002; 46:2972 - 2982
- Bird LA, Peh CA, Kollnberger S, Elliott T, McMichael AJ, Bowness P. Lymphoblastoid cells express HLA-B27 homodimers both intracellularly and at the cell surface following endosomal recycling. Eur J Immunol 2003; 33:748 - 759
- Allen RL, Trowsdale J. Recognition of classical and heavy chain forms of HLA-B27 by leukocyte receptors. Curr Mol Med 2004; 4:59 - 65
- Kollnberger S, Bird LA, Roddis M, Hacquard-Bouder C, Kubagawa H, Bodmer HC, et al. HLA-B27 heavy chain homodimers are expressed in HLA-B27 transgenic rodent models of spondyloarthritis and are ligands for paired Ig-like receptors. J Immunol 2004; 173:1699 - 1710
- Mear JP, Schreiber KL, Münz C, Zhu X, Stevanovic S, Rammensee HG, et al. Misfolding of HLA-B27 as a result of its B pocket suggests a novel mechanism for its role in susceptibility to spondyloarthropathies. J Immunol 1999; 163:6665 - 6670
- Dangoria NS, DeLay ML, Kingsbury DJ, Mear JP, Uchanska-Ziegler B, Ziegler A, et al. HLA-B27 misfolding is associated with aberrant intermolecular disulfide bond formation (dimerization) in the endoplasmic reticulum. J Biol Chem 2002; 277:23459 - 23468
- Colbert RA. HLA-B27 misfolding: A solution to the spondyloarthropathy conundrum?. Mol Med Today 2000; 6:224 - 230
- Turner MJ, Sowders DP, DeLay ML, Mohapatra R, Bai S, Smith JA, et al. HLA-B27 misfolding in transgenic rats is associated with activation of the unfolded protein response. J Immunol 2005; 175:2438 - 2448
- Turner MJ, Delay ML, Bai S, Klenk E, Colbert RA. HLA-B27 upregulation causes accumulation of misfolded heavy chains and correlates with the magnitude of the unfolded protein response in transgenic rats: Implications for the pathogenesis of spondylarthritis-like disease. Arthritis Rheum 2007; 56:215 - 223
- Laitio P, Virtala M, Salmi M, Pelliniemi LJ, Yu DT, Granfors K. HLA-B27 modulates intracellular survival of salmonella enteritidis in human monocytic cells. Eur J Immunol 1997; 27:1331 - 1338
- Virtala M, Kirveskari J, Granfors K. HLA-B27 modulates the survival of salmonella enteritidis in transfected L cells, possibly by impaired nitric oxide production. Infect Immun 1997; 65:4236 - 4242
- Penttinen MA, Heiskanen KM, Mohapatra R, DeLay ML, Colbert RA, Sistonen L, et al. Enhanced intracellular replication of Salmonella enteritidis in HLA-B27-expressing human monocytic cells: Dependency on glutamic acid at position 45 in the B pocket of HLA-B27. Arthritis Rheum 2004; 50:2255 - 2263
- Sahlberg AS, Penttinen MA, Heiskanen KM, Colbert RA, Sistonen L, Granfors K. Evidence that the p38 MAP kinase pathway is dysregulated in HLA-B27-expressing human monocytic cells: Correlation with HLA-B27 misfolding. Arthritis Rheum 2007; 56:2652 - 2662
- Penttinen MA, Ekman PGranfors K. Non-antigen presenting effects of HLA-B27. Curr Mol Med 2004; 4:41 - 49
- Bridges JP, Xu Y, Na CL, Wong HR, Weaver TE. Adaptation and increased susceptibility to infection associated with constitutive expression of misfolded SP-C. J Cell Biol 2006; 172:395 - 407
- Penttinen MA, Holmberg CI, Sistonen L, Granfors K. HLA-B27 modulates nuclear factor κB activation in human monocytic cells exposed to lipopolysaccharide. Arthritis Rheum 2002; 46:2172 - 2180
- Gregersen N, Bross P, Vang S, Christensen JH. Protein Misfolding and Human Disease. Annu Rev Genomics Hum Genet 2006; 7:103 - 124
- Hammer GE, Kanaseki T, Shastri N. The final touches make perfect the peptide-MHC class I repertoire. Immunity 2007; 26:397 - 406
- Kienast A, Preuss M, Winkler M, Dick TP. Redox regulation of peptide receptivity of major histocompatibility complex class I molecules by ERp57 and tapasin. Nat Immunol 2007; 8:864 - 872
- Bouvier M. Accessory proteins and the assembly of human class I MHC molecules: a molecular and structural perspective. Mol Immunol 2003; 39:697 - 706
- Dick TP. Assembly of MHC class I peptide complexes from the perspective of disulfide bond formation. Cell Mol Life Sci 2004; 61:547 - 556
- Peh CA, Burrows SR, Barnden M, Khanna R, Cresswell P, Moss DJ, et al. HLA-B27-restricted antigen presentation in the absence of tapasin reveals polymorphism in mechanisms of HLA class I peptide loading. Immunity 1998; 8:531 - 542
- Purcell AW, Gorman JJ, Garcia-Peydró M, Paradela A, Burrows SR, Talbo GH, et al. Quantitative and qualitative influences of tapasin on the class I peptide repertoire. J Immunol 2001; 166:1016 - 1027
- Dick TP, Bangia N, Peaper DR, Cresswell P. Disulfide bond isomerization and the assembly of MHC class I-peptide complexes. Immunity 2002; 16:87 - 98
- Serwold T, Gaw S, Shastri N. ER aminopeptidases generate a unique pool of peptides for MHC class I molecules. Nat Immunol 2001; 2:644 - 651
- Serwold T, Gonzalez F, Kim J, Jacob R, Shastri N. ERAAP customizes peptides for MHC class I molecules in the endoplasmic reticulum. Nature 2002; 419:480 - 483
- Hammer GE, Gonzalez F, Champsaur M, Cado D, Shastri N. The aminopeptidase ERAAP shapes the peptide repertoire displayed by major histocompatibility complex class I molecules. Nat Immunol 2006; 7:103 - 112
- York IA, Brehm MA, Zendzian S, Towne CF, Rock KL. Endoplasmic reticulum aminopeptidase 1 (ERAP1) trims MHC class I-presented peptides in vivo and plays an important role in immunodominance. Proc Natl Acad Sci USA 2006; 103:9202 - 9207
- Kanaseki T, Blanchard N, Hammer GE, Gonzalez F, Shastri N. ERAAP synergizes with MHC class I molecules to make the final cut in the antigenic peptide precursors in the endoplasmic reticulum. Immunity 2006; 25:795 - 806
- Hammer GE, Gonzalez F, James E, Nolla H, Shastri N. In the absence of aminopeptidase ERAAP, MHC class I molecules present many unstable and highly immunogenic peptides. Nat Immunol 2007; 8:101 - 108
- Cui X, Hawari F, Alsaaty S, Lawrence M, Combs CA, Geng W, et al. Identification of ARTS-1 as a novel TNFR1-binding protein that promotes TNFR1 ectodomain shedding. J Clin Invest 2002; 110:515 - 526
- Cui X, Rouhani FN, Hawari F, Levine SJ. Shedding of the type II IL-1 decoy receptor requires a multifunctional aminopeptidase, aminopeptidase regulator of TNF receptor type 1 shedding. J Immunol 2003; 171:6814 - 6819
- Cui X, Rouhani FN, Hawari F, Levine SJ. An aminopeptidase, ARTS-1, is required for interleukin-6 receptor shedding. J Biol Chem 2003; 278:28677 - 28685
- Schroder M, Kaufman RJ. The mammalian unfolded protein response. Annu Rev Biochem 2005; 74:739 - 789
- Perlmutter DH. Liver injury in alpha1-antitrypsin deficiency: An aggregated protein induces mitochondrial injury. J Clin Invest 2002; 110:1579 - 1583
- Iwakoshi NN, Lee AH, Vallabhajosyula P, Otipoby KL, Rajewsky K, Glimcher LH. Plasma cell differentiation and the unfolded protein response intersect at the transcription factor XBP-1. Nat Immunol 2003; 4:321 - 329
- Oyadomari S, Araki E, Mori M. Endoplasmic reticulum stress-mediated apoptosis in pancreatic beta-cells. Apoptosis 2002; 7:335 - 345
- Ron D. Proteotoxicity in the endoplasmic reticulum: Lessons from the Akita diabetic mouse. J Clin Invest 2002; 109:443 - 445
- Southwood CM, Garbern J, Jiang W, Gow A. The unfolded protein response modulates disease severity in Pelizaeus-Merzbacher disease. Neuron 2002; 36:585 - 596
- Nagaraju K, Casciola-Rosen L, Lundberg I, Rawat R, Cutting S, Thapliyal R, et al. Activation of the endoplasmic reticulum stress response in autoimmune myositis: potential role in muscle fiber damage and dysfunction. Arthritis Rheum 2005; 52:1824 - 1835
- Griffin TA, Reed AM. Pathogenesis of myositis in children. Curr Opin Rheumatol 2007; 19:487 - 491
- Nagaraju K, Raben N, Loeffler L, Parker T, Rochon PJ, Lee E, et al. Conditional upregulation of MHC class I in skeletal muscle leads to self-sustaining autoimmune myositis and myositis-specific autoantibodies. Proc Natl Acad Sci USA 2000; 97:9209 - 9214
- Colbert RA, Rowland-Jones SL, McMichael AJ, Frelinger JA. Allele-specific B pocket transplant in class I major histocompatibility complex protein changes requirement for anchor residue at P2 of peptide. Proc Natl Acad Sci USA 1993; 90:6879 - 6883
- Meusser B, Hirsch C, Jarosch E, Sommer T. ERAD: the long road to destruction. Nat Cell Biol 2005; 7:766 - 772
- Tran TM, Satumtira N, Dorris ML, May E, Wang A, Furuta E, et al. HLA-B27 in transgenic rats forms disulfide-linked heavy chain oligomers and multimers that bind to the chaperone BiP. J Immunol 2004; 172:5110 - 5119
- Antoniou AN, Ford S, Taurog JD, Butcher GW, Powis SJ. Formation of HLA-B27 homodimers and their relationship to assembly kinetics. J Biol Chem 2004; 279:8895 - 8902
- Colbert RA. The immunobiology of HLA-B27: variations on a theme. Curr Mol Med 2004; 4:21 - 30
- Whelan MA, Archer JR. Chemical reactivity of an HLA-B27 thiol group. Eur J Immunol 1993; 23:3278 - 3285
- Kostyu DD, Hannick LI, Traweek JL, Ghanayem M, Heilpern D, Dawson DV. HLA class I polymorphism: Structure and function and still questions. Hum Immunol 1997; 57:1 - 18
- Breban M, Hammer RE, Richardson JA, Taurog JD. Transfer of the inflammatory disease of HLA-B27 transgenic rats by bone marrow engraftment. J Exp Med 1993; 178:1607 - 1616
- Breban M, Fernández-Sueiro JL, Richardson JA, Hadavand RR, Maika SD, Hammer RE, et al. T-cells, but not thymic exposure to HLA-B27, are required for the inflammatory disease of HLA-B27 transgenic rats. J Immunol 1996; 156:794 - 803
- May E, Dorris ML, Satumtira N, Iqbal I, Rehman MI, Lightfoot E, et al. CD8ab T-cells are not essential to the pathogenesis of arthritis or colitis in HLA-B27 transgenic rats. J Immunol 2003; 170:1099 - 1105
- Rath HC, Herfarth HH, Ikeda JS, Grenther WB, Hamm TE Jr, Balish E, et al. Normal luminal bacteria, especially bacteroides species, mediate chronic colitis, gastritis and arthritis in HLA-B27/human β2 microglobulin transgenic rats. J Clin Invest 1996; 98:945 - 953
- Rath HC, Wilson KH, Sartor RB. Differential induction of colitis and gastritis in HLA-B27 transgenic rats selectively colonized with Bacteroides vulgatus or Escherichia coli. Infect Immun 1999; 67:2969 - 2974
- Boyle LH, Goodall JC, Opat SS, Gaston JS. The recognition of HLA-B27 by human CD4+ T-lymphocytes. J Immunol 2001; 167:2619 - 2624
- Boyle LH, Goodall JC, Gaston JS. Major histocompatibility complex class I-restricted alloreactive CD4+ T-cells. Immunology 2004; 112:54 - 63
- Roddis M, Carter RW, Sun MY, Weissensteiner T, McMichael AJ, Bowness P, et al. Fully functional HLA B27-restricted CD4+ as well as CD8+ T-cell responses in TCR transgenic mice. J Immunol 2004; 172:155 - 161
- Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol 2007; 8:519 - 529
- Lin W, Harding HP, Ron D, Popko B. Endoplasmic reticulum stress modulates the response of myelinating oligodendrocytes to the immune cytokine interferon-gamma. J Cell Biol 2005; 169:603 - 612
- Smith JA, Turner MJ, DeLay ML, Kleck EI, Sowders DP, Colbert RA. Endoplasmic reticulum stress-induced and the unfolded protein response are linked to synergistic IFNβ induction via X-box binding protein-1. Eur J Immunol 2008; 38:1194 - 1203
- Lee AH, Iwakoshi NN, Glimcher LH. XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol Cell Biol 2003; 23:7448 - 7459
- Taniguchi T, Takaoka A. A weak signal for strong responses: Interferon-alpha/beta revisited. Nat Rev Mol Cell Biol 2001; 2:378 - 386
- Montoya M, Schiavoni G, Mattei F, Gresser I, Belardelli F, Borrow P, et al. Type I interferons produced by dendritic cells promote their phenotypic and functional activation. Blood 2002; 99:3263 - 3271
- Gautier G, Humbert M, Deauvieau F, Scuiller M, Hiscott J, Bates EE, et al. A type I interferon autocrine-paracrine loop is involved in Toll-like receptor-induced interleukin-12p70 secretion by dendritic cells. J Exp Med 2005; 201:1435 - 1446
- Seimon TA, Obstfeld A, Moore KJ, Golenbock DT, Tabas I. Combinatorial pattern recognition receptor signaling alters the balance of life and death in macrophages. Proc Natl Acad Sci USA 2006; 103:19794 - 19799
- Li Y, Schwabe RF, DeVries-Seimon T, Yao PM, Gerbod-Giannone MC, Tall AR, et al. Free cholesterol-loaded macrophages are an abundant source of tumor necrosis factoralpha and interleukin-6: model of NFkappaB and map kinase-dependent inflammation in advanced atherosclerosis. J Biol Chem 2005; 280:21763 - 21772
- Colbert RA, Turner MJ, DeLay ML, Smith JA, Klenk EI, Sowders DP, et al. HLA-B27 misfolding activates the Il-23/Il-17 axis via the unfolded protein response in transgenic rats: evidence for a novel mechanism of inflammation. Arth Rheum 2007; 54:515
- Mosmann TR, Coffman RL. TH1 and TH2 cells: Different patterns of lymphokine secretion lead to different functional properties. Annu Rev Immunol 1989; 7:145 - 173
- Bottomly K. A functional dichotomy in CD4+ T-lymphocytes. Immunol Today 1988; 9:268 - 274
- Langrish CL, McKenzie BS, Wilson NJ, de Waal Malefyt R, Kastelein RA, Cua DJ. IL-12 and IL-23: Master regulators of innate and adaptive immunity. Immunol Rev 2004; 202:96 - 105
- Harrington LE, Hatton RD, Mangan PR, Turner H, Murphy TL, Murphy KM, et al. Interleukin 17-producing CD4+ effector T-cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol 2005; 6:1123 - 1132
- Park H, Li Z, Yang XO, Chang SH, Nurieva R, Wang YH, et al. A distinct lineage of CD4 T-cells regulates tissue inflammation by producing interleukin 17. Nat Immunol 2005; 6:1133 - 1141
- Weaver CT, Hatton RD, Mangan PR, Harrington LE. IL-17 family cytokines and the expanding diversity of effector T-cell lineages. Annu Rev Immunol 2007; 25:821 - 852
- Cua DJ, Sherlock J, Chen Y, Murphy CA, Joyce B, Seymour B, et al. Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature 2003; 421:744 - 748
- Chen Y, Langrish CL, McKenzie B, Joyce-Shaikh B, Stumhofer JS, McClanahan T, et al. Anti-IL-23 therapy inhibits multiple inflammatory pathways and ameliorates autoimmune encephalomyelitis. J Clin Invest 2006; 116:1317 - 1326
- Yen D, Cheung J, Scheerens H, Poulet F, McClanahan T, McKenzie B, et al. IL-23 is essential for T-cell-mediated colitis and promotes inflammation via IL-17 and IL-6. J Clin Invest 2006; 116:1310 - 1316
- Uhlig HH, McKenzie BS, Hue S, Thompson C, Joyce-Shaikh B, Stepankova R, et al. Differential activity of IL-12 and IL-23 in mucosal and systemic innate immune pathology. Immunity 2006; 25:309 - 318
- Zheng Y, Danilenko DM, Valdez P, Kasman I, Eastham-Anderson J, Wu J, et al. Interleukin-22, a T(H)17 cytokine, mediates IL-23-induced dermal inflammation and acanthosis. Nature 2007; 445:648 - 651
- Chen CH, Lin KC, Yu DT, Yang C, Huang F, Chen HA, et al. Serum matrix metalloproteinases and tissue inhibitors of metalloproteinases in ankylosing spondylitis: MMP-3 is a reproducibly sensitive and specific biomarker of disease activity. Rheumatology (Oxford) 2006; 45:414 - 420
- Nakae S, Nambu A, Sudo K, Iwakura Y. Suppression of immune induction of collagen-induced arthritis in IL-17-deficient mice. J Immunol 2003; 171:6173 - 6177
- Kolls JK, Linden A. Interleukin-17 family members and inflammation. Immunity 2004; 21:467 - 476
- Mangan PR, Harrington LE, O'Quinn DB, Helms WS, Bullard DC, Elson CO, et al. Transforming growth factor-beta induces development of the T(H)17 lineage. Nature 2006; 441:231 - 234
- McGeachy MJ, Bak-Jensen KS, Chen Y, Tato CM, Blumenschein W, McClanahan T, et al. TGFbeta and IL-6 drive the production of IL-17 and IL-10 by T-cells and restrain T(H)-17 cell-mediated pathology. Nat Immunol 2007; 8:1390 - 1397
- Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, et al. The orphan nuclear receptor RORgt directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell 2006; 126:1121 - 1133
- Aggarwal S, Ghilardi N, Xie MH, de Sauvage FJ, Gurney AL. Interleukin-23 promotes a distinct CD4 T-cell activation state characterized by the production of interleukin-17. J Biol Chem 2003; 278:1910 - 1914
- Langrish CL, Chen Y, Blumenschein WM, Mattson J, Basham B, Sedgwick JD, et al. IL-23 drives a pathogenic T-cell population that induces autoimmune inflammation. J Exp Med 2005; 201:233 - 240
- Kastelein RA, Hunter CA, Cua DJ. Discovery and biology of IL-23 and IL-27: related but functionally distinct regulators of inflammation. Annu Rev Immunol 2007; 25:221 - 242
- Annunziato F, Cosmi L, Santarlasci V, Maggi L, Liotta F, Mazzinghi B, et al. Phenotypic and functional features of human Th17 cells. J Exp Med 2007; 204:1849 - 1861
- Wilson NJ, Boniface K, Chan JR, McKenzie BS, Blumenschein WM, Mattson JD, et al. Development, cytokine profile and function of human interleukin 17-producing helper T-cells. Nat Immunol 2007; 8:950 - 957
- Wiekowski MT, Leach MW, Evans EW, Sullivan L, Chen SC, Vassileva G, et al. Ubiquitous transgenic expression of the IL-23 subunit p19 induces multiorgan inflammation, runting, infertility and premature death. J Immunol 2001; 166:7563 - 7570
- Biron CA, Nguyen KB, Pien GC, Cousens LP, Salazar-Mather TP. Natural killer cells in antiviral defense: Function and regulation by innate cytokines. Annu Rev Immunol 1999; 17:189 - 220
- Taurog JD. The mystery of HLA-B27: if it isn't one thing, it's another. Arthritis Rheum 2007; 56:2478 - 2481
- Olivieri I, Padula A, Cianco G, Moro L, Durante E, Guadiano C, et al. The HLA-B* 2709 subtype in a patient with undifferentiated spondarthritis. Ann Rheum Dis 2000; 59:654 - 655
- Olivieri I, Ciancio G, Padula A, Gaudiano C, Masciandaro S, Moro L, et al. The B*2709 subtype does not give absolute protection against spondyloarthropathy. Arthritis Rheum 2000; 43:265
- Olivieri I, D'Angelo S, Scarano E, Santospirito V, Padula A. The HLA-B*2709 subtype in a woman with early ankylosing spondylitis. Arthritis Rheum 2007; 56:2805 - 2807
- Cauli A, Vacca A, Mameli A, Passiu G, Fiorillo MT, Sorrentino R, et al. A Sardinian patient with ankylosing spondylitis and HLA-B*2709 co-occurring with HLA-B*1403. Arthritis Rheum 2007; 56:2807 - 2809
- Fiorillo MT, Cauli A, Carcassi C, Bitti PP, Vacca A, Passiu G, et al. Two distinctive HLA haplotypes harbor the B27 alleles negatively or positively associated with ankylosing spondylitis in Sardinia: implications for pathogenesis. Arth Rheum 2003; 48:1385 - 1389
- Taurog JD. HLA-DR4 and the spondyloarthropathies. Ann Rheum Dis 2002; 61:193 - 194
- López-Larrea C, Sujirachato K, Mehra NK, Chiewsilp P, Isarangkura D, Kanga U, et al. HLA-B27 subtypes in Asian patients with ankylosing spondylitis: evidence for new associations. Tissue Antigens 1995; 45:169 - 176
- Gonzalez-Roces S, Alvarez MV, Gonzalez S, Dieye A, Makni H, Woodfield DG, et al. HLA-B27 polymorphism and worldwide susceptibility to ankylosing spondylitis. Tissue Antigens 1997; 49:116 - 123
- Hou TY, Chen HC, Chen CH, Chang DM, Liu FC, Lai JH. Usefulness of human leucocyte antigen-B27 subtypes in predicting ankylosing spondylitis: Taiwan experience. Intern Med J 2007; 37:749 - 752
- Duerr RH, Taylor KD, Brant SR, Rioux JD, Silverberg MS, Daly MJ, et al. A genomewide association study identifies IL23R as an inflammatory bowel disease gene. Science 2006; 314:1461 - 1463
- Cargill M, Schrodi SJ, Chang M, Garcia VE, Brandon R, Callis KP, et al. A large-scale genetic association study confirms IL12B and leads to the identification of IL23R as psoriasis-risk genes. Am J Hum Genet 2007; 80:273 - 290