Abstract
The structural organization of the amyloidogenic β-proteins containing 40 amino acid residues (Aβ40) was studied by the high temperature molecular dynamics simulations in the acidic (pH~3) and basic (pH~8) pH regions. The obtained data suggest that the central Ala21-Gly29 segment of Aβ40, can adopt folded and partially unfolded structures. At the basic pH, this segment forms folded structures, stabilized by electrostatic interactions and hydrogen bonds. At the acidic pH, it forms partially unfolded structures. Two other segments flanking to the central segment exhibit the propensity to adopt unstable inter-converting α-helical, 310-helical and turn-like structures. One of these segments is comprised of the Ala30-Val36 residues at both of the considered pHs. The second segment is comprised of the Glu11-Phe20 at the basic pH and of the Glu11-Val24 residues at the acidic pHs. The revealed pH-dependent structuration of the Aβ40 allowed us to suggest a possible scenario for initial Aβ aggregation. According to this scenario, the occurrence of the partially unfolded states of the Ala21-Gly29 segment plays main role in the Aβ oligomerization process.
Introduction
Amyloidogenic, natively unfolded β-protein (amyloid-β or Aβ), consisting of 40 or 42 amino acid residues (referred as Aβ40 or Aβ42, correspondingly), is the major component of the amyloid deposits in the brains of patients with Alzheimer's disease (AD).Citation1,Citation2 This protein can form self-aggregated complexes containing fibrilar and non-fibrilar structures that are neurotoxic and responsible for AD pathogenesis.Citation3–Citation5 A better understanding of the formation of the amyloid oligomers and protofibril is crucial for the rational design of new therapeutic strategies to prevent Aβ aggregation into neurotoxic structures and, perhaps, to treat AD.
Solid state NMR experimentsCitation6–Citation9 suggest that a “cross-β supra-molecular” structure of Aβ fibrils is formed in vitro from a core of the elongated β-sheets. Attempts to find structural reorganizations of the Aβ monomers that precede their folding into the predominantly β-stranded structures did not reveal a single pathway.Citation10–Citation30 These studies have shown that, the Aβ monomers may contain the α-helix, β-strand or collapsed-coil structural components, as well as a mixture of the α-helix/β-strand or a-helix/collapsed-coil structural components at the early stages of aggregation. Recent worksCitation31–Citation34 suggest that partial unfolding of the somehow initially folded Aβ(21–30) segment may be necessary for the subsequent fibrilization of the Aβ monomers. Overall, the existing experimental data suggest that the Aβ monomers should undergo structural reorganization events to approach an intermediate structural state required for the initiation the Aβ oligomeric and protofibril aggregation. The mechanisms of these structural reorganizations have not been fully discovered yet.
In this work, we have focused on studying how the pH of the surrounding media can influence the structural organization of Aβ monomers. The rationale behind this work is based on an in vitro observation showing that the oligomeric and protofibril aggregation of the Aβ protein can be induced by the pH and temperature changes.Citation13,Citation35–Citation39 It was demonstratedCitation13,Citation38,Citation39 that the aggregation rate of the Aβ fibril formation is much faster at the acidic pH compared to the neutral and basic pHs. Data obtained by solid state NMR spectroscopyCitation9 and atomic force microscopy experimentsCitation40–Citation42 also showed pH-dependent structural alterations of the Aβ monomers. On the other hand, pH values determine protonated and deprotonated states of the amino acid residues having ionogenic groups. Since there are many residues with the ionogenic groups in the Aβ sequence, the pH changes can shift the balance of the Aβ intra-protein electrostatic interactions that, in turn, can change the structural propensities of the monomers and alter the process of Aβ oligomerization and fibrilization.
To elucidate the role of electrostatic interactions in the structural organization of Aβ monomers, we performed the high temperature (HT) molecular dynamics (MD) simulations of the Aβ40 monomer at the acidic (pH ∼ 3) and basic (pH ∼ 8) pHs, which were referred to as pH 3 and pH 8, correspondingly. The HT simulations are used to increase conformational sampling and to diminish the probabilities that a peptide or unstructured short protein will be trapped in one or a few local-minimum-energy states. The HT molecular dynamic simulation protocols were found to be useful in many studies.Citation43–Citation49
By combining the HT simulations data with known experimental data, we have proposed a model of the pH-dependent structural organization of the Aβ monomer. This model suggests that electrostatic interactions within the central Ala21-Gly29 segment play a crucial role in the ability of Aβ40 to commence or escape oligomerization and fibrilization processes. The proposed model provides an explanation of some experimental data on the Aβ oligomerization and fibrilization. The model also clarifies the nature of the structural changes caused by several well-known pathological mutations of the Aβ proteins and sheds light on a mechanism that can trigger the initiation of fibril formation.
Results and Discussions
To estimate a general dynamic behavior of the Aβ40 structure during the time of the full simulations, the radii of gyrations (Rg) were calculated using coordinates of the heavy backbone atoms (Cα, C, N) of the whole Aβ40 presented in the MDA3, MDA8, MDB3 and MDB8 trajectory files (see Methods). The dynamics behavior of the corresponding values of the Rg (RgA3, RgA8, RgB3 and RgB8, respectively) is shown in and B. As can be seen from , the RgA8 values are changing during a rather short time from the starting values of ∼15 Å to the average value, <RgA8> ≈ 11.6 ± 1.8 Å (after ∼150 ps), while the RgB8 are started from the values of ∼35 Å and approach (after about 2 ns) very similar values, <RgB8> ≈ 11.5 ± 2.0 Å. A similar picture was observed for the behavior of the Aβ40 at pH 3 (). Starting from the values of ∼15 Å and ∼35 Å, the corresponding RgA3 and RgB3 also approach very similar average values: <RgA3> ≈ 10.7 ± 1.2 Å (after ∼300 ps) and <RgB3> ≈ 10.75 ± 1.3 Å (after ∼4 ns).
Therefore, during the MD simulations, the average values of the radius of gyration of the Aβ40 monomer are dependent on the considered pHs, but not from the choice of the initial structure. Thus, the structural behavior of Aβ40 can be viewed as a behavior of a flexible, unstable system, adopting several pH-dependent structural formations ().
To analyze the pH-dependent structural formations of Aβ40, the detailed structural analysis of the conformers, selected at the last 10 ns of the performed simulations, was carried out. For this purpose, data collected in the RSA8, RSA3, RSB8 and RSB3 files (see Methods) were used. Analysis of the formation of secondary structure elements (SSE) for RSA3 and RSA8 as well as RSB3 and RSB8 structural sets showed that at both pHs the amino acids of the Aβ40 are mainly involved in the formation of α-helixes, 310-helixes and β-turns and random coils ( and ). It should be noted that the hydrogen bonded β-strand structure, which was absent in the considered structural sets, and two other structures, isolated β-strand and π-helix, that appeared very rarely in these sets, were omitted from this table. To analyze whether the structural propensity (SP) of the amino acid residues of Aβ40 depend on initial starting structures (A and B) during MD simulations, we calculated the correlation coefficients (R) between SP of amino acid residues within several segments of the Aβ40 sequence to be involved in the formation of the α-helix, 310-helix and β-turn structures as well as the random coil obtained for two initial structures at the same pH ( and ). We found that during the last 10 ns of the A and B runs (see Methods) the corresponding amino acid residues within the Glu11-Val39 segment have rather strongly correlated propensity to form the α-helix, 310-helix and random coil structural elements: the correlation coefficients, R, are 0.92, 0.86 and 0.95 (respectively) at pH 8 and 0.87, 0.91 and 0.86 at pH 3. Smaller, but remarkable correlations between the SP of these residues to belong to the β-turn structures were also observed (R = 0.73 at pH 8 and R = 0.6 at pH 3). This analysis suggests that, during the last 10 ns of MD simulations at the given pH, the residues within the Glu11-Val39 segment for both runs have similar tendency to be involved in helix- and coil-like structural motifs. This result suggests that the amino acid residues from the Glu11-Val39 sequence at the given pH exhibit similar structural behavior independent from the initial structures.
In contrast to the Glu11-Val39 segment, the residues within the Ala2-Tyr10 segment exhibited low or moderate ability (data are not shown) to be involved in the formation of the α-helix, 310-helix and random coil at pH 3 (R = 0.6, 0.86 and 0.64, respectively). At pH 8 such a correlation is absent (R = −0.3, −0.4 and −0.1, respectively). The similarities in adopting the β-turn structures were found to also be absent at pH 8 (R = 0.1) and low at pH 3 (R = 0.6). Thus, we concluded that the structural preferences of the Ala2-Tyr10 segment significantly depend not only on pH, but also on the initial structure. Because the first 10 residues in Aβ40 were found to be structurally disordered in the fibrillesCitation6,Citation8 and are likely not important for aggregation,Citation8 we did not perform any additional analysis of this segment.
presents the SP of the amino acid residues of the Aβ40 at the last 10 ns of the MD simulations performed for A and B initial structures at pH 8. This figure suggests the presence of three distinct zones exhibiting different SP along the Aβ40 core domain (Glu11-Val36). These three zones are formed from the following residues: Glu11-Phe20 (Zone I), Ala21-Gly29 (Zone II) and Ala30-Val36 (Zone III). At both pHs, the residues within Zones I and III demonstrate propensity to be involved in the formation of a-helix, 310-helix and β-turn structural motifs. This is in contrast to Zone II, where the residues at pH 8 do not exhibit a significant propensity to be involved in forming any of the considered types of secondary structures.
Structural preferences of the amino acid residues within Zone I and Zone III.
As can be seen from , and , the SP of the amino acid residues within Zone I (Glu11-Phe20) and Zone III (Ala30-Val36) of the Aβ40 core domain can be described as inter-converting structural formations. These formations include helical, β-turn and random coil structural motifs at both pH 3 and pH 8. To better understand how the choice of the initial structure influences the structural preferences of the amino acid residues within Zone I and Zone III at the considered pH, we performed the structural cluster analysis described below. This analysis was carried out using the RS8 and RS3 datasets, which were obtained by combining the RSA8 and RSB8 as well as RSA3 and RSB3 sets taken at pH 8 and pH 3, correspondingly. Thus, within each of the combined sets, RS8 and RS3, 1,000 MD generated conformers were analyzed. We performed structural cluster analyses of the following five tetrapeptide segments: Glu11-His14, His14-Leu17 and Leu17-Phe20 as well as Ala30-Gly33 and Gly33-Val36. In this analysis, the value of RMSD ≤ 1.0 Å (for the heavy backbone atoms) for the segments was taken as a criterion of a structural similarity (see Methods). To test whether the average structures of the clusters obtained under the high temperature MD simulations are persistent at low temperatures, these structures were energy-minimized and compared (by RMSD calculated for the backbone heavy atoms) with the “ideal” α-helical, 310-helical, π-helical and β-strand structures (as described in Methods). The results of these comparisons are summarized in . Data in this table suggest that at the given pH the most preferable helix-like structuration within segments belonging to Zone I and Zone III, is independent of the choice of the initial structure (or this dependence is very weak). The helical structuration of the core domain found in the present work is consistent with the results of MD simulations performed at room temperature for the Aβ(10–35) peptide in aqueous solution at the neutral pH.Citation61 In that work it was shown that the Aβ(10–35) peptide predominantly adopts helix-like structures, while the propensities to adopt β-strand-like structures were much weaker. Helical formations of the Aβ monomer and its fragments at neutral and low pHs were also found in other works.Citation10,Citation17,Citation20,Citation24,Citation30,Citation62
It should be noted that a general tendency of the peptide segments within Zone I and Zone III to adopt the helix-like formations is only weakly dependent on the considered pHs (–). These data taken together with the data obtained at neutral pHs in the aforementioned works suggest that the protonation or deprotonation of the Aβ histidines (His6, His13 and His14) apparently do not change the general tendency of the residues within Zones I and III to preferably adopt helical conformations.
Structural preferences of the central Zone II comprised of the Ala21-Gly29 residues.
As can be seen from and , the structural preferences of the central Ala21-Gly29 segment are pH dependent. To better understand how pH changes influence the structural preferences of this segment we performed a cluster analysis of four structural sets, RSA8, RSA3, RSB8 and RSB3, corresponding to A and B initial starting structures. Each average structure of the obtained structural clusters of the Ala21-Gly29 segment was energy-minimized using the procedure described in Methods. We found (see below) that the structural shapes of the segment obtained at the high temperature are persistent after the energy minimization and are very similar for both initial structures. Therefore, we only present data obtained for the initial structure B in .
The performed analysis suggests that, at pH 8, almost all of the structures formed by the residues belonging to the Ala21-Gly29 segment can be assigned to four clusters shown in . As can be seen from this table, cluster 1 is comprised of about 84% of the considered structures, while three other clusters are low populated. The minimized and non-minimized average structures had RMSD ∼1.0 Å for the heavy backbone atoms. The Cα-trace of the energy-minimized average structure corresponding to the most populated cluster is shown in . As can be seen from this figure, the Ala21-Val24 residues of this segment form the hook-shaped structures, while the Gly25-Gly29 residues form the bend-shaped structures. Overall, the fold of the whole Ala21-Gly29 segment resembles a distorted omega-like (Ω) structure. We called the state in which the Ala21-Gly29 segment forms compact, Ω-shaped structures, the stable (or folded) state. The states in which the corresponding residues form structures grouped into the low populated clusters (, clusters 2, 3 and 4), we called the meta-stable states of the Ala21-Gly29 segment.
The structures formed by the Ala21-Gly29 segment at pH 3 are grouped in many relatively low populated clusters (). None of these clusters contains the majority of the corresponding structures. Therefore, at this pH, the Ala21-Gly29 segment exists in several meta-stable states, and does not exhibit the propensity to be in a stable state. The structures in the meta-stable states differ significantly from those in the stable state. Two factors are responsible for this. First, the Ala21-Val24 part of the central segment exhibits the propensity to adopt the α-, 310-helix and β-turn-like structures (with the RMSDs equal to 0.53, 0.47 and 0.6 Å, respectively). The second factor is that the orientations of the Ala21-Val24 structures relative to the bend-shaped structures typical for the Gly25-Gly29 segment are significantly different from the orientation taking place in the Ω-shaped structures. Internal conformational flexibility of the central segment at the Gly25 position is associated with the varieties of its orientations taking place in the meta-stable states.
It is important to note that in addition to the aforementioned differences in structural preferences exhibited by the residues belonging to the Ala21-Gly29 segment, there are also some commonalities in the structural behavior of this segment at pH 8 and pH 3. In fact, at both pHs, the Gly25-Gly29 residues adopt similar bend-folded structures. As a part of these folded structures, the Gly25-Lys28 residues form the turn-like structures resembling the β-turn I type structuresCitation63 with a smaller distortion at pH 3 and a larger distortion at pH 8. At both pHs, there is also a similarity between the secondary structure elements adopted by the Ala21-Val24 residues in meta-stable states (for example, see clusters 4, 5, 6 and 8 in ). Further, at both pHs the Gly29 residue exhibits some conformational flexibility. Finally, at the considered pHs, remarkable conformation flexibility of the central segment at the Gly25 position was also found in the meta-stable states.
Overall, the performed analysis suggests that at pH 8 the central Ala21-Gly29 segment prefers to be in the stable state, in which it adopts the compactly-folded, Ω-shaped structures. In rare cases, however, this segment can also be observed in the partially unfolded, meta-stable states (see ). In these states the Ala21-Val24 residues adopt β-strand- and helix-like structures different from the hook-shaped structures adopted by these residues, when the central segment is in the folded state. At pH 3, the central segment exists in meta-stable, partially-unfolded states, in which the Ala21-Val24 residues prefer to adopt α-, 310-helix and turn-like structures. In the folded and partially-unfolded states, the Gly25-Gly29 residues form similar bend-shaped structures.
The revealed structural behavior of the central Ala21-Gly29 segment of the Aβ proteins can explain the pH-related dependences of the attractive forces between two Aβ proteins brought in close proximity in atomic force microscopy (AFM) experiments.Citation40,Citation42 These experiments have shown a non-linear, sigmoidal functional relationship between the attractive forces and pH magnitudes. These forces were found to be minimal in the wide range of the basic pH and increased significantly at the acidic pH. This can be interpreted in the following way. Our MD simulations data showed that at pH 3 the central Ala21-Gly29 segment of Aβ40 forms the partially unfolded structures and has a flexible structure associated with inter-conversions of α-, 310-helix and β-turn structural modes (). At pH 8, however, this segment prefers to be in the stable state, in which it adopts the compactly-folded, Ω-shaped structures. Thus, at pH 3 the Aβ core domain exhibits a more flexible structuration compared to its relatively rigged, Ω-shaped structure at pH 8. It is clear that when Aβ proteins are brought together in close proximity in enough random orientation, one relative to another, they have a greater ability to form a larger number of intermolecular contacts, when each of these molecules has higher conformational flexibility. Such an effect is consistent with a flexible protein binding mechanism manifested by the fly-casting effect.Citation64 Thus, the flexible structuration of the Aβ at pH 3 in conjunction with the hydrophobicity effectCitation65 induces further collapse of a larger number of the hydrophobic residues of the interacting proteins and promotes their hydrophobic assembly. Therefore, we suggested that an increase of the attractive forces between two Aβ proteins measured by the AFM at the acidic pH is due to increasing the number of the inter-protein hydrophobic contacts at this pH compared to the ones at the basic pH. In other words, the increase of the attractive forces between two Aβ proteins measured by AFM at the acidic pH compared to basic pHCitation40,Citation42 reflects the difference in the inter-protein hydrophobic interactions at these pHs.
Physicochemical nature of the folded state of the Ala21-Gly29 segment.
To determine the features of forming the Ω-shaped structures, we analyzed the possible nets of hydrogen bonds (H-bonds) and the residue-residue contacts within the Ala21-Gly29 segment at pH 8. For this purpose, the MDB8 and MDA8 trajectory files were utilized. The 6.5 Å cutoff distances between the centers of mass of the side chains were used as the criterion for finding the contacting residues.Citation66 The nets of H-bonds and sets of the residue-residue contacts stabilizing the Ω-shaped structures at pH 8 were found to be very similar in both runs. Therefore, below we present the results obtained by the analysis of only one trajectory file, namely MDB8.
Our analysis showed that when the Ω-shaped structures are formed, the side-chains of the Lys28 and Glu22 residues, as well as the Lys28 and Asp23 residues are close to each other. At pH 8 these structures are stabilized by the network of the H-bonds, formed between the ionogenic groups of Glu22 and Lys28, as well as between the ionogenic group of Asp23 and backbone NH groups of the Val24-Gly29 segment ( and ). In some cases, the H-bonds between the ionogenic group of Asp23 and the polar OH group of the Ser26 side-chain were also observed (). Our analysis of the distances between the heavy atoms (CD and NZ) of the Glu22 and Lys28 side-chains in the MD8 trajectory showed that around 70% of MD time this distance was less than 7.0 Å. This suggested that at pH 8, even without the Glu22-Lys28 salt-bridge formation, the long-range ion pairCitation67 between the ionogenic groups of these residues takes place and facilitates the formation of the Ω-shaped structures. It should be noted that the revealed network of the H-bonds and the formation of the Glu22-Lys28 ion pair are well consistent with the data obtained by replica exchange MD simulations performed for the Aβ(21–30) and Aβ(10–35) fragments at the neutral pH.Citation29,Citation34 Thus, our data, taken together with data obtained by Baumketner et al.Citation29,Citation34 suggest that formation of the Ω-shaped structures can be adopted by the Ala21-Gly29 segment at pH 8 as well as at neutral pHs.
Overall, our data suggested that the favorable electrostatic interactions within the Glu22-Gly29 segment play a critical role in forming and stabilizing the O-shaped structures. This conclusion is strongly supported by a comparative analysis of the structural preferences of the Ala21-Gly29 segment during the MD simulations performed at both pHs. At pH 8 (as well as at the neutral pH, when the Glu22 and Asp23 residues are deprotonated and negatively charged), the Ala21-Gly29 segment adopts well-stabilized, O-shaped structures. At pH 3 (when the Glu22 and Asp23 residues are protonated and uncharged) the Ω-shaped structures are not formed. It should be noted that the destruction of the Ω-shaped structure and high flexibility of the Ala21-Gly29 segment at pH 3 are associated with the protonation of the Glu22 and Asp23 residues at this pH. The revealed structural flexibility of the Ala21-Gly29 segment is well consistent with the data obtained by Massi et al.Citation61 In that work it was shown that the Glu22Gln mutation in the Aβ(10–35) fragment causes larger structural flexibility of the mutated fragment at the neutral pH and room temperature when compared with the structural flexibility of the wild-type fragment. The important role of the electrostatic interactions of the Glu22, Asp23 and Lys28 residues shown in our work are also consistent with data of the recent work carried out for the Aβ(21–30) peptide at the neutral pH.Citation68
The revealed dependence of the structural propensities of the Ala21-Gly29 segment on intra-segmental electrostatic interactions explains some structural effects caused by several pathological mutations of the Aβ protein at the neutral pH: the Arctic [Glu22Gly], Dutch [Glu22Gln], Italian [Glu22Lys] and Iowa [Asp23Asn]. In fact, these mutations suppress or disturb favorable electrostatic interactions within the Ala21-Gly29 segment and thus impede the formation of the Ω-shaped structures. Therefore, instead of forming the stable and relatively rigid Ω-shaped structures, the central segment adopts partially unfolded states that can initiate the subsequent structural changes. This assumption is consistent with the observations that the structural changes caused by the aforementioned pathological mutations increase the rate of nucleation and enhance the Aβ fibrilization, occurring at the neutral pH.Citation69–Citation73 On the other hand, our assumption on the electrostatic nature of the forming Ω-shaped structure is well correlated with data recently observed by Grant et al.Citation74 suggesting that all disease-associated amino acid substitutions at Glu22 or Asp23 positions destabilize the turn-like structure of the Glu21-Ala30 segment of the Aβ protein that, in turn, causes familial forms of AD and cerebral amyloid angiopathy.
Thus, our data, taken together with the recent literature data, allowed us to suggest that any neutralization (by pH changing or mutations) of the charged aspartic (Glu22) and/or glutamic (Asp23) acid residues in the Aβ monomer can induce a partial unfolding of the Ala21-Gly29 segment and facilitate the amyloid oligomer and protofibril formations.
Role of structural organization of the Aβ40 core domain in the oligomerization and protofibril aggregation of the Aβ monomers.
Our analysis showed that during MD simulations performed for two different initial structures (A—the structure with a high α-helix content and B—a polyproline II structure) the conformational evolution of the Aβ40 core domain achieves rather steady pH-dependent states, exhibiting flexible, unstable helix-like structuration.
, and point to the existence of three structural zones within the Aβ40 core domain. Amino acid residues within Zones I and III prefer to form flexible and fluctuating helix-like structures (molten helixes) at both pHs, while the residues within Zone II have pronounced propensities to adopt rather stable structures at pH 8. Based on their structural properties, we called Zones I and III the molten (or transit) helical zones and Zone II the folded zone. The length of Zone I depends on pH; at pH 8 this zone is comprised of the Glu11-Phe20 residues, while at pH 3 this zone includes the Glu11-Val24 residues. The length of Zone II is also pH-dependent; at pH 8 this zone includes the Ala21-Gly29 residues, while at pH 3 it includes only the Gly25-Gly29 residues. The length of Zone III is pH-independent and includes the Ala30-Val36 residues. The pH-dependent structuration of the Aβ40 core domain associated with the aforementioned three zones is schematically presented in . Below we discuss the roles that the length and structural properties of these zones can play in the processes of oligomerization and protofibril aggregation of the Aβ monomers.
The performed cluster analysis has shown that the nine residue segment, Ala21-Gly29, prefers to form the compactly-folded, Ω-shaped structures at pH 8, while at pH 3 the shorter, five-residue segment, Gly25-Gly29, can adopt relatively rigid, bend-like structures. Interestingly, at pH 3 (and in rare cases at pH 8) the Ala21-Val24 segment becomes unfolded and exhibits propensities to form fluctuating structures and thus can be considered as a continuation of the molten helical Zone I. These observations can be correlated with the experimentally observed different abilities of the Aβ monomers to aggregate into oligomeric and protofibril entities at different pHs. In fact, in vitro experiments have shown that the oligomeric and protofibril aggregation of the Aβ monomers can be induced by low pH and elevated temperature.Citation13,Citation35–Citation39 The aggregation rate of the Aβ fibril formation is much faster at the acidic pH compared to the neutral pH.Citation13,Citation38,Citation39 By combining our computational data with the known experimental data, we suggested that the protofibril aggregation of the Aβ proteins is associated with the diminishing length of the folded zone and with unfolding of the Ala21-Val24 segment. On the other hand, the formation of the compactly-folded, Ω-shaped structures by the Ala21-Gly29 residues is detrimental to the Aβ oligomerization.
Therefore, we suggested that the length (five, but not nine residues) and structure (bend-shaped, but not Ω-shaped) of the folded zone contribute to the protofibril aggregation of the Aβ proteins. The agreement between our model of the Aβ fibrilization and known experimental dataCitation6,Citation8,Citation9 can be further enhanced by the assumption that the flexible, unstable helix-like structures formed by the residues from the molten Zones I and III could be transformed into β-strand structures, when the Aβ monomers form fibrils. This assumption is consistent with the concept of “unstable helix” state of the Aβ proteinCitation10 and with the hypothesis11 that considers a “discordant” α-helix of the Lys16-Asp23 segment as strongly favorable for β-strand formation. Our assumption is also based on the solid-state NMR data,Citation6,Citation8,Citation9 which suggest that the Aβ40 fibrilization is associated with the formation of β-strand structures within Glu11-Val24 and Ala30-Val40 segments. These β-strand structures are bridged by the Gly25-Gly29 segment, which adopts a bent structure.
Below we explain how fluctuating structures, adopted by the Glu11-Val24 and Ala30-Val36 residues, can be transformed into the β-strand structures. Let us assume that the process of this structural conversion has two phases. In the first phase, suitable fluctuating structures, adopted by the Glu11-Val24 and Ala30-Val36 residues, bridged by the Gly25-Gly29 segment, can occasionally form structural motifs resembling helical hairpins. The formation of such motifs is facilitated by: (1) the inherent preferences of the Glu11-Val24 and Ala30-Val36 segments to form fluctuating (molten) helix-like structural motifs; (2) the presence of a remarkable number of the hydrophobic residues within these motifs; (3) the occurrence of the bend-shaped structures adopted by the Gly25-Gly29 segment; and (4) the presence of internal conformational variability of the Aβ40 at the Gly25 and Gly29 positions. In the second phase, when the occasionally formed hairpins are hydrophobically collapsed, the transition of α-helices into β-strands can occur.
The proposed hydrophobic mechanism can be illustrated by the cylindrical diagram presented in . As can be seen from this diagram, the distribution of the hydrophobic residues along two cylinders, presenting α-helices formed by the Glu11-Val24 and Val30-Val36 residues, is typical for the cases when α-helices connected by an inter-helical loop are complementarily packed.Citation75–Citation77 The character of the continuous hydrophobic clusters on the helical surfaces and length of the gap (Gly25-Gly29) between these clusters suggest that the hydrophobic residues of these α-helices have the possibility to be packed in a face-to-face fashion and, in such a manner, to form structures reminding helical hairpins.
According to this diagram, the continuous hydrophobic clusters can be formed on three sides of the helix, adopted by the Val30-Val36 segment, and on two sides of the helix, adopted by the Glu11-Val24 segment. Moreover, the seven-amino acid sequence, comprised of the Leu17-Val18-Phe19-Phe20-Ala21-Glu22-Asp23 residues (see Helix 1 on the ), has a hydrophobic pattern, typical for the coiled coil structural motif.Citation78,Citation79 In this motif, the first and fourth positions (in our case, the Leu17 and Phe20 residues) must be occupied by the hydrophobic residues. These residues form the hydrophobic core of the super coil structure assembled from parallel or anti- parallel α-helices.Citation78,Citation79 Recently, it was found that the fifth and seventh residues (in our case, the Ala21 and Asp23) can have non-polar side chains.Citation79 Thus, one can suggest that the coiled coil motif formed by the Leu17-Asp23 segment within the Aβ monomer and hydrophobic effect (providing direct contact of the corresponding hydrophobic clusters) are important elements of the Aβ oligomerization process. Such a process should involve the formation of the super coil structures, built from the parallel oriented helical segments comprised of the Leu17-Asp23 residues of several Aβ monomers. In this case, the Asp23 has to be buried in the hydrophobic environment. At pH 3, when Asp23 is in the protonated state, this should not be a problem. At pH 8 (as well as at the neutral pH ∼7), this is also possible when the Asp23 ionogenic group can form an ion pair with the ionogenic group of Lys28.
Through the diffusion-collision process of the Aβ monomers, the hydrophobic clusters can provide a mutual basis necessary for Aβ monomers to initiate oligomerization, driven by the hydrophobic interactions.Citation18,Citation80 This process can be followed by a transition of α-helices into β-strands forming the major structural motifs (β-sheets) typical for Aβ fibrils.Citation6,Citation8,Citation9,Citation12–Citation15,Citation22 Transition of the helical structures into the β-strand structures was suggested in several works.Citation10–Citation13,Citation15,Citation61 One of the possible reasons why the helical structures can be altered into the β-strand structures is discussed below.
Due to the oligomerization of hydrophobically collapsed Aβ40 monomers, the residues involved in the helical formations can become significantly isolated from the aqueous solvent and exposed to the protein-core-like medium. It may be reasonable to suggest that this new environment can promote the conversion of the unstable flexible α-helical structures into β-strands. The possible physicochemical basis for this conversion may be associated with alterations in a balance of intra-residual non-covalent (electrostatic) interactions (that determine conformational propensities of an amino acid residue in protein) when the polar (solvent) surroundings are replaced by a non-polar medium typical for the internal protein region.Citation81–Citation83 This effect assumes that amino acid residues buried within the protein-like media can have different preferences to adopt possible conformations compared to the conformational preferences of these residues when they are exposed to solvent at the protein-solvent interface. Our previous dataCitation82 suggest that amino acid residues located in the interior of proteins can have higher preferences to adopt extended, β-strand-like conformations compared to the residues located on the protein surface.
The proposed model of the Aβ oligomerization enriches the existing structural models of Aβ oligomerization and fibrillization. For example, one of the advanced structural models was suggested by Fezoui et al.Citation84,Citation85 for the de novo designed 38-residue helical hairpin peptide. When incubated in aqueous solvent at the neutral pH, this peptide can form the protease resistant fibrils that have the morphologic, structural and tinctorial properties of authentic amyloid fibrils formed by Aβ40 and Aβ42.Citation86 In that work it was found that the partially folded intermediates, formed by the de novo designed peptide, containing the α-helix-turn-α-helix (αtα) motif, can mediate the initial stages of fibril formation. Our model suggests that the molten helical hairpins, occasionally adopted by the residues of the Aβ40 core domain, can initiate the Aβ fibril formation, followed by a transition of the helical zones into the β-strands typical for the Aβ fibrils. In addition, the proposed model addresses why and how the molten helical structures are transferred into the β-strand structures. Interestingly, the way these transient zones are marked within the Aβ40 sequence (by coding the amino acid sequence of the Glu11-Val24 and Ala30-Val36 segments, having inherent conformational propensities to adopt unstable fluctuating helix-like structures) is consistent with one observed in other short, intrinsically unfolded proteins.Citation87
Methods
MD simulations protocol.
The MD simulations of the Aβ40 protein were performed starting from two initial structures (starting  points). The first initial structure (referred to as the A structure) was taken as a representative from the family of Aβ40 structures derived from the NMR experiments;Citation11 namely, it was the tenth structure of the 1BA4 file from the Brookhaven Data Bank. This structure suggests that the Asp1-His14 segment of Aβ40 is unstructured, while the Asp15-Val36 segment adopts α-helical conformation with a kink at Gly25-Asn27 residues. The second initial structure (referred to as the B structure) was taken with the backbone dihedral angles for each amino acid residue adopting the values (φ ≈ −69°, Ψ ≈ 149°); these dihedral angles are characteristic for the polyproline II structure (PPII). The PPII was chosen as the starting structure because all 20 types of amino acid residues can adopt this conformationCitation50,Citation51 and PPII can be easily transformed into other types of secondary structures.Citation51 The MD simulations performed starting from the A and B initial structures we called the A and B runs, respectively.
points). The first initial structure (referred to as the A structure) was taken as a representative from the family of Aβ40 structures derived from the NMR experiments;Citation11 namely, it was the tenth structure of the 1BA4 file from the Brookhaven Data Bank. This structure suggests that the Asp1-His14 segment of Aβ40 is unstructured, while the Asp15-Val36 segment adopts α-helical conformation with a kink at Gly25-Asn27 residues. The second initial structure (referred to as the B structure) was taken with the backbone dihedral angles for each amino acid residue adopting the values (φ ≈ −69°, Ψ ≈ 149°); these dihedral angles are characteristic for the polyproline II structure (PPII). The PPII was chosen as the starting structure because all 20 types of amino acid residues can adopt this conformationCitation50,Citation51 and PPII can be easily transformed into other types of secondary structures.Citation51 The MD simulations performed starting from the A and B initial structures we called the A and B runs, respectively.
The structural behavior of Aβ40 was studied at pH 3 and pH 8. At pH 8 all aspartic and glutamic acids in the Aβ40 were considered in deprotonated states, while at pH 3 these amino acid residues and histidines were protonated. All lysine and arginine residues of the Aβ40 were protonated at both pHs. Thus, at pH 8 the Glu and Asp residues were negatively charged, Arg and Lys residues were positively charged, and His had a zero charge. At the pH 3, the Glu and Asp residues were considered non-charged, whereas Arg, Lys and His residues were positively charged.
The SANDER module of the AMBER 7 package with an all-atom force field ff99 was used for the MD simulations.Citation52,Citation53 The LEaP module of this packageCitation53 was utilized to protonate/deprotonate the aforementioned residues. The initial Aβ40 structures were solvated in a periodic box of the TIP3P model water.Citation54 The box size was chosen to provide not less than 9.0 Å from each wall to any atom of Aβ40.
All simulations were performed using periodic boundary conditions and the SHAKE algorithmCitation55 at a constant temperature of 500° K using the Berendsen coupling algorithm (NTT = 1).Citation56 The constant pressure MD simulations (NTP protocol)Citation53 were performed with the 1 fs time step. To carry out the simulations at pH 8, positive counter ions, Na+, were added at the most negative regions of the electrostatic potential to neutralize the simulation system. It was done by mutually replacing the corresponding water molecules. Analogously, the negative counter ions, Cl−, were used to neutralize the simulation system at pH 3.
The electrostatic interactions were calculated by the Particle Mesh Ewald methodCitation57 implemented in SANDER. The van der Waals and electrostatic interactions were evaluated with a 9.0 Å cutoff value. The non-bonded pair list was updated every 100 steps. After the initial 1,000 steps of energy minimization, the system was equilibrated during 100 ps of the MD run with positional constraints. During the first 20 ps of the equilibration, the system was heated from 0 to 500° K. The positional constraints were gradually reduced from 50 to 0.5 kcal/(mol*Å) within the equilibration time.
The productive (without positional constraints) MD simulations were carried out at 10.9 ns and 30 ns for the A and B initial structures, respectively. Data generated during MD simulations were stored in trajectory files every 1 ps, providing 10,900 and 30,000 conformers for the A and B runs, respectively. Two trajectory files (MDA3 and MDA8) were generated during the A runs at pH 3 and pH 8, respectively. Analogously, the MDB3 and MDB8 files were generated during the B runs at pH 3 and pH 8, respectively. Four representative structural sets, RSA3 and RSA8 as well as RSB3 and RSB8, were obtained from the corresponding MDA3, MDA8, MDB3 and MDB8 trajectory files by extracting 500 structures evenly distributed during the last 10 ns of the MD simulations.
All simulations were performed on the Beowulf computer cluster containing 32x nodes Atipa Dual-Xeon (32x Xeon 3.06 GHz and 32x Xeon 2.4 GHz). Data analysis was carried out on the SGI O2 workstation. The collected data were analyzed by the CARNAL module of AMBER as well as by the PROTABLE module and the graphic tools of the SYBYL software package (Tripos, Inc.).
Structural propensities (SP).
The involvement of each residue in the formation of secondary structure elements (SSE) was estimated by the Kabsh-Sander procedureCitation58 incorporated in the SYBYL software package. This procedure allows one to assign the involvement of each amino acid residue in forming α-helix (H), isolated β-strand (B), hydrogen bonded β-strand (E), 310-helix (G), π-helix (I) and hydrogen bonded β-turn (T). Residues unassigned to any of the aforementioned SSE were considered to be in a random coil (C). The Kabsh-Sander procedure was performed for the RSA8, RSA3 and RSB8, RSB3 sets to calculate SSE profiles along the Aβ40 amino acid sequence for both A and B runs carried out at both considered pHs. The occupation percentages (or structural propensities) of the seven structural states (H, G, I, B, E, T and C) for each amino acid residue of Aβ40 were also calculated.
Structural cluster analysis.
The RSA3 and RSA8 as well as RSB3 and RSB8 sets were analyzed to determine the possible structural organization of the different segments of the Aβ40 sequence. For this purpose, a procedure for cluster analysis similar to one described earlierCitation59,Citation60 was used.
Structural cluster analysis was performed for several Aβ40 segments. For each cluster (comprising all conformers satisfying a given RMSD criterion), the average structure was calculated by the SYBYL software package. These average structures were energy-minimized to test the persistence of these structures at low temperatures. The minimization was performed by the SYBYL software package using the AMBER7 FF99 force field, distance-dependent dielectric function ϵ = 2r, and a gradient convergence value of 0.05 kcal/(mol Å) for termination of the minimization process. The minimized structures were compared (by RMSD calculated for the backbone heavy atoms) with the “ideal” α-helical, 310-helical, π-helical and β-strand structures. These “ideal” structures were set up by the backbone dihedral angles for each amino acid residue: φ = −57°, Ψ = −47° (for α-helix); φ = −50°, Ψ = −28° (for 310-helix); φ = −57°, Ψ = −70° (for π-helix); and φ = −139°, Ψ = 135° (for β-strand).
Hydrogen bond analysis.
The analysis of the hydrogen bonds (H-bonds) was carried out for the Glu22-Gly29 segment using the set of structures collected in the MDA8 and MDB8 trajectory file. This analysis was performed by the CARNAL module of the AMBER with the use of the 4.0 Å cut-off distances between the corresponding heavy atoms (CD for Glu22, CG for Asp23, NH and NZ for Lys28, NH and OG for Ser26 as well as NH for Val24, Gly25, Asn27 and Gly29) and the cut-off donor-hydrogenacceptor angle within 60° of linearity.
Conclusion
In this work, the pH-dependent structural features of the Aβ40 core domain were determined by the high temperature MD simulations, performed at pH ∼8 and pH ∼3. The obtained data combined with the known experimental data allowed us to describe the structural properties of the Aβ40 monomers and enrich existing models of Aβ oligomerization and fibrillization.
Analysis of the MD simulations data suggests the existence of three structural zones within the Aβ40 core domain: two molten zones (Zones I and III) and one folded zone (Zone II). At both considered pHs the amino acid residues within Zones I and III form flexible and fluctuating helix-like structures (molten helixes). In contrast, the amino acid residues within Zone II form rather stable structures at pH 8. The length of Zone I depends on the pH. At pH 8, this zone is comprised of the Glu11-Phe20 residues, while at pH 3 this zone includes the Glu11-Val24 residues. The length of Zone II is also pH dependent. At pH 8 this zone is comprised of nine residues (Ala21-Gly29) and at pH 3 it includes only five residues (Gly25-Gly29). At pH 3 (and in rare cases at pH 8), the Ala21-Val24 residues form fluctuating structures and thus can be considered as a part of the molten helical Zone I. The length of Zone III appears to be pH-independent and includes the Ala30-Val36 residues.
Combined analysis of the MD simulations and experimental data suggested that adopting the compactly-folded, Ω-shaped structures by the nine residue segment, Ala21-Gly29, blocks the Aβ oligomerization process. When this segment becomes partially unfolded (i.e., when the Ala21-Val24 residues form fluctuating structures) and when only five residues (Gly25-Gly29) of this segment are folded in the bend-like structures, the processes of Aβ oligomerization and fibrillization can occur.
Analysis of the flexible helical structures, formed within the molten zones (Zone I and Zone III), revealed the existence of the continuous hydrophobic clusters, which can be responsible for intra-molecular hydrophobic collapsing of the molten zones. In addition to this, we suggested that the formation of helical structures by the Leu17-Asp23 residues can be instrumental in inter-molecular hydrophobic collapsing of Aβ monomers and forming the super-coil structures, built from the parallel oriented helical segments that include the Leu17-Asp23 residues of several Aβ monomers. This conclusion is based on the fact that the Leu17-Asp23 segment has a hydrophobic pattern typical for the coiled coil structures. Our results suggested that helical structuration of the Leu17-Asp23 residues is possible when the Ala21-Gly29 segment is partially unfolded and the Ala21-Val24 residues from this segment form fluctuating helical structures. Finally, we described a physicochemical mechanism of how the hydrophobic collapsing can cause a transition of the molten helical zones into the β-strands typical for the Aβ fibrils.
The details of the Aβ40 structural organization described in this work and the proposed model of the Aβ oligomerization and fibrillization will be useful for further studies of possible mechanisms of the Aβ aggregation.
Figures and Tables
Figure 1 Dynamics behavior of the radii of gyration (Rg) of Aβ40 during MD simulations performed for (A and B) initial structures at both considered pHs. Notes: Plots of the radii of gyration calculated (in Angstrom) for backbone atoms (Cα, C, N) of Aβ40 at pH 8 and pH 3 are shown in the upper and lower parts of the figure, correspondingly. Colored curves correspond to the A (magenta) and B (blue) initial structures (see Methods).
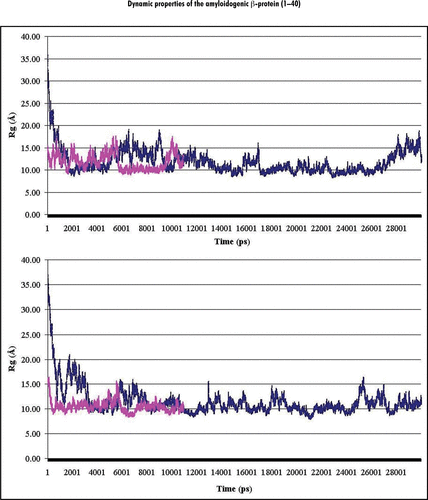
Figure 2 Percentage (%) of involvement of the amino acid residues within the Aβ40 sequence in forming the secondary structural elements (structural propensity) during the last 10 ns of MD simulations at pH 8: (A and B) present secondary structure sets determined by the analyses of the RSA8 and RSB8 structural sets, correspondingly. Notes: The secondary structure elements were depicted by different colors: α-helix (green), 310-helix (blue), β-turn (yellow) and random coil (red).
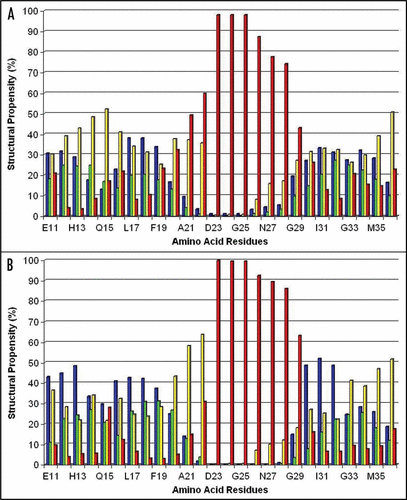
Figure 3 The trace of the Cα-atoms of the average Ω-shaped structure formed by the Ala21-Gly29 segment in the stable state at pH 8.
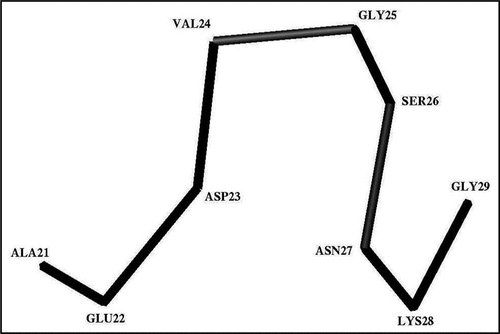
Figure 4 A typical three-dimensional structure formed by the Glu22-Gly29 segment in the stable state at pH 8. Notes: Observed H-bonds between polar groups are shown by dash-lines. CD, CG, OG and NZ note the heavy atoms of the donor-acceptor groups of the interacting residues.
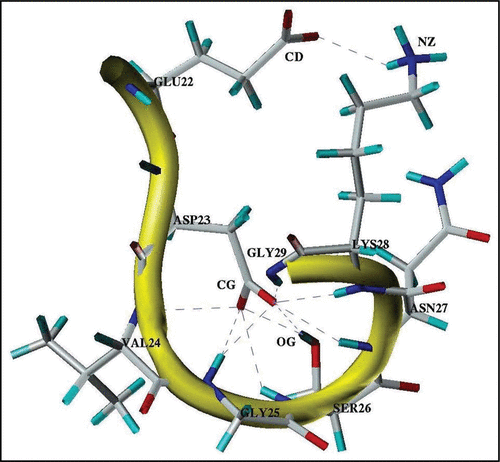
Figure 5 Schematic presentation of the major structural states of the Glu11-Val36 segment of the Aβ40 core domain considered at pH 8 and pH 3. The rectangular blocks along the sequence designate the flexible and fluctuating helix-like structural modes: Glu11-Phe20 (Helix 1′) at pH 8; Glu11-Val24 (Helix 1) at pH 3, and Ala30-Val36 (Helix 2) at both pHs. The Ala21-Gly29 residues at pH 8 form the Ω-shaped structure (comprised of about 84% of the MD generated structures). The Gly25-Gly29 residues at both pHs form a bend-shaped structure.
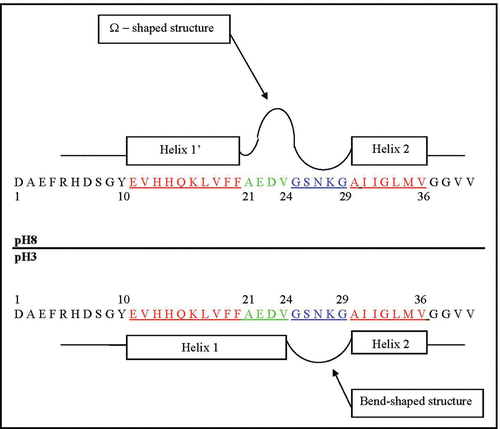
Figure 6 Helical net layout of the Glu11-Gly37 segment. Notes: The sequence is presented by the net diagram typical for two α-helices, formed by the Glu11-Val24 (Helix 1) and Ala30-Val36 (Helix 2) segments. The residues forming the continuous hydrophobic clusters are marked by dark circles. The residues forming the bend structures (Gly25-Gly29) are marked by the hexagons. The numbers present the positions of the corresponding amino acid residues in the Aβ40 sequence.
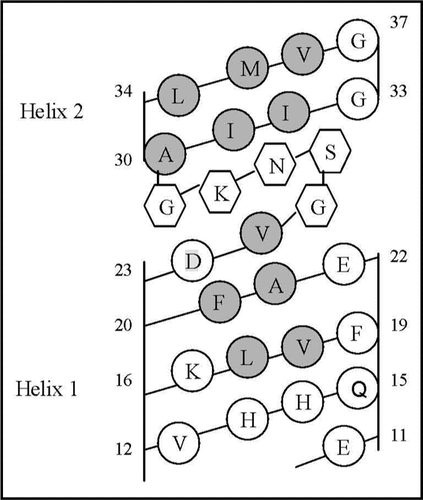
Table 1 The structural propensities (occupation percentage) of amino acid residues within Glu11-Val39 segment to be involved in the formation of secondary structure elements revealed from the RSA8 and RSA3 structural sets at the last 10 ns of the MD simulations
Table 2 The structural propensities (occupation percentage) of amino acid residues within Glu11-Val39 segment to be involved in the formation of secondary structure elements revealed from the RSB8 and RSB3 structural sets at the last 10 ns of the MD simulations RSA8 RSA3
Table 3 Percentage (%) of helical structures adopted by five tetrapeptide segments of Aβ40 at pH 8 and pH 3
Table 4 The percentage (%) of structures assigned to the most populated clusters formed by the Ala21-Gly29 segment and overall description of the structures formed by the Ala21-Val24 residues
Table 5 Percentage of the cases (%), in which the hydrogen bonds (H-bonds) between the donor-acceptor groups of the residues within the Glu22-Gly29 segment are formed
Acknowledgements
We thank Dr. L. Kinarsky (University Nebraska Medical Center) and Dr. V. Uversky (Indiana University) for helpful remarks. We acknowledge the use of the UNMC Bioinformatics Core Facility partially supported by NRI Nebraska Research Initiative, NIH Cancer Center Support Grant, NIH BRIN Nebraska Training Network in Functional Genomics and NIH grant 1-PN1 EY016593-01.
References
- Hardy J, Higgins GA. Alzheimer's disease: the amyloid cascade hypothesis. Science 1992; 256:184 - 185
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science 2002; 297:353 - 356
- Kirkitadze MD, Bitan G, Teplow DB. Paradigm shifts in Alzheimer's disease and other neurodegenerative disorders: the emerging role of oligomeric assemblies. J Neurosci Res 2002; 69:567 - 577
- Lacor PN, Buniel MC, Chang L, Fernandez SJ, Gong Y, Viola KL, et al. Synaptic targeting by Alzheimer's-related amyloid β oligomer. J Neurosci 2004; 24:10191 - 10200
- Petkova AT, Leapman RD, Guo Z, Yau W-M, Mattson MP, Tycko R. Self-propagating, molecular-level polymorphism in Altzheimer's β-amyloid fibrils. Science 2005; 307:262 - 265
- Tycko R. Insights into the amyloid folding problem from solid-state NMR. Biochem 2003; 42:3151 - 3159
- Tycko R. Progress towards a molecular-level structural understanding of amyloid fibrils. Curr Opin Struct Biology 2004; 14:96 - 103
- Petkova AT, Ishii Y, Balbach JJ, Antzutkin ON, Leapman RD, Delaglio F, et al. A structural model for Alzheimer's beta-amyloid fibrils based on experimental constraints from solid state NMR. Proc Natl Acad Sci USA 2002; 99:16742 - 16747
- Petkova AT, Yau W-M, Tycko R. Experimental constraints on quaternary structure in Alzheimer's β-amyloid fibrils. Biochem 2006; 45:498 - 512
- Chen YR, Huang HB, Chyan L, Shiao MS, Lin TH, Chen YC. The effect of Aβ conformation on the metal affinity and aggregation mechanism studied by circular dichroism spectroscopy. J Biochem 2006; 139:733 - 740
- Paivio A, Nordling E, Kallberg Y, Thyberg J, Jahansson J. Stabilization of discordant helices in amyloid fibil-forming proteins. Protei Sci 2004; 13:1251 - 1259
- Walsh DM, Hartley DM, Kusumoto Y, Fezoui Y, Condron MM, Lomakin A, et al. Amyloid β-protein fibrillogenesis. J Biolog Chem 1999; 274:25945 - 25952
- Kirkitadze MD, Condron MM, Teplow DB. Identification and characterization of key kinetic intermediates in Amyloid β-protein fibrillogenesis. J Mol Biol 2001; 312:1103 - 1119
- Massi F, Peng JW, Lee JP, Straub JE. Simulation study of the structure and dynamics of the Alzheimer's amyloid peptide congener in solution. Biophys J 2001; 80:31 - 44
- Klimov DK, Thirumalai D. Dissecting the assembly of Aβ16–22 amyloid peptide into antiparallel β sheets. Structure 2003; 11:295 - 307
- Klimov DK, Straub JE, Thirumalai D. Aqueous urea solution destabilizes Aβ16-22 oligomers. Proc Natl Acad Sci USA 2004; 101:14760 - 14765
- Urbanc B, Cruz L, Ding F, Sammond D, Khare S, Buldyrev SV, et al. Molecular dynamics simulation of amyloid β dimer formation. Biophys J 2004; 87:2310 - 2321
- Urbanc B, Cruz L, Teplow DB, Stanley HE. Computer simulations of Alzheimer's amyloid β-protein folding and assembly. Curr Alzheimer Res 2006; 3:493 - 504
- Daidone I, Simona F, Roccatano D, Broglia RA, Tiana G, Colombo G, et al. β-Hairpin conformation of fibrillogenic proteins: structure and a-β transition mechanism revealed by molecular dynamics simulations. Proteins 2004; 57:198 - 204
- Xu Y, Shen J, Luo X, Zhu W, Chen K, Maand J, Jiang H. Conformational transition of amyloid β-peptide. Proc Natl Acad Sci USA 2005; 102:5403 - 5407
- Han W, Wu YD. A strand-loop-strand structure is a possible intermediate in fibril elongation: long time simulations of amyloid-beta peptide (10–35). J Am Chem Soc 2005; 127:15408 - 15416
- Buchete N-V, Tycko R, Hummer G. Molecular dynamics simulations of Alzheimer's β-amyloid protofilaments. J Mol Biol 2005; 353:804 - 821
- Ma B, Nussinov R. Simulations as analytical tools to understand protein aggregation and predict amyloid conformation. Curr Opin Chem Biol 2006; 10:445 - 452
- Flöck D, Colacino S, Colombo G, Di Nola A. Misfolding of the amyloid β-protein: a molecular dynamics study. Proteins 2006; 62:183 - 192
- Baumketner A, Bernstein SL, Wyttenbach T, Bitan G, Teplow DB, Bowers MT, et al. Amyloid β-protein monomer structure: a computational and experimental study. Protein Sci 2006; 15:420 - 428
- Wei G, Shea J-E. Effects of solvent on the structure of the Alzheimer amyloid-β (25–35) peptide. Biophys J 2006; 91:1638 - 1647
- Teplow DB, Lazo ND, Bitan G, Bernstein S, Wyttenbach T, Bowers MT, et al. Elucidating amyloid β-protein folding and assembly: a multidisciplinary approach. Acc Chem Res 2006; 39:635 - 645
- Yun S, Urbanc B, Cruz L, Bitan G, Teplow DB, Stanley HE. Role of electrostatic interactions in amyloid β-protein (Aβ) oligomer formation: a discrete molecular dynamics study. Biophys J 2007; 92:4064 - 4077
- Baumketner A, Shea J-E. The structure of the Alzheimer amyloid β 10–35 peptide probed throght replica-exchange molecular dynamics simulations in explicit solvent. J Mol Biol 2007; 366:275 - 285
- Khandogin J, Brooks CL III. Linking folding with aggregation in Alzheimer's β-amyloid peptides. Proc Natl Acad Sci USA 2007; 104:16880 - 16885
- Lazo ND, Grant MA, Condron MC, Rigby AC, Teplow DB. On the nucleation of amyloid β-protein monomer folding. Protein Sci 2005; 14:1581 - 1596
- Borreguero JM, Urbanc B, Lazo ND, Buldyrev SV, Teplow DB, Stanley HE. Folding events in the 21–30 region of amyloid β-protein (Aβ) studied in silico. Proc Natl Acad Sci USA 2005; 102:6015 - 6020
- Cruz L, Urbanc B, Borreguero JM, Lazo ND, Teplow DB, Stanley HE. Solvent and mutation effects on the nucleation of amyloid β-protein folding. Proc Natl Acad Sci USA 2005; 102:18258 - 18263
- Baumketner A, Bernstein SL, Wyttenbach T, Lazo ND, Teplow DB, Bowers MT, Shea J-E. Structure of the 21–30 fragment of amyloid β-protein. Protein Sci 2006; 15:1239 - 1247
- Hirakura Y, Lin M-C, Kagan BL. Alzheimar amyloid Aβ1–42 channels: effects of solvent, pH and Congo Red. J Neurosci Res 1999; 57:458 - 466
- Gursky O, Aleshkov S. Temperature-dependent beta-sheet formation in beta-amyloid Abeta(1–40) peptide in water: uncoupling beta-structure folding from aggregation. Biochim Biophys Acta 2000; 1476:93 - 102
- Chu HL, Lin SY. Temperature-induced conformational changes in amyloid beta(1–40) peptide investigated by simultaneous FT-IR microspectroscopy with thermal system. Biophys Chem 2001; 89:173 - 180
- Stine WB Jr, Dahlgren KN, Kraff GA, LaDu MJ. In vitro characterization of conditions for amyloid-β peptide oligomerization and fibrillogenesis. J Biol Chem 2003; 278:11612 - 11622
- Hortschansky P, Schroeckh V, Christopeit T, Zandomeneghi G, Fandrich M. The aggregation kinetics of Alzheimer's beta-amyloid peptide is controlled by stochastic nucleation. Protein Sci 2005; 14:1753 - 1759
- McAllister C, Karymov MA, Kawano Y, Lushnikov AY, Mikheikin A, Uversky VN, Lyubchenko YL. Protein interactions and misfolding analyzed by AFM force spectroscopy. J Mol Biol 2005; 354:1028 - 1042
- Krasnoslobodtsev AV, Shlyakhtenko LS, Ukraintsev E, Zaikova TO, Keana JFW, Lyubchenko YL. Nanomedicine and protein misfolding diseases. Nanomedicine 2005; 1:300 - 305
- Lyubchenko YL, Sherman S, Shlyakhtenko LS, Uversky VN. Nanoimaging for protein misfolding and related diseases. J Cell Biochem 2006; 99:53 - 70
- Daggett V, Levitt M. Realistic simulations of native-protein dynamics in solution and beyond. Annu Rev Biophys Biomol Struct 1993; 22:353 - 380
- Mohanty D, Elber R, Thirumalai D, Beglov D, Roux B. Kinetics of peptide folding: computer simulations of SYPFDV and peptide variants in water. J Mol Biol 1997; 272:423 - 442
- Duan Y, Kollman PA. Pathways to a protein folding intermediate observed in a 1-micro-second simulation in aqueous solution. Science 1998; 282:740 - 744
- Daura X, van Gunsteren WF, Makr AE. Folding-unfolding thermodynamics of a β-heptapeptide from equilibrium simulations. Proteins 1999; 34:269 - 280
- Daggett V. Long timescale simulations. Curr Opin Struct Biol 2000; 10:160 - 164
- Beck DAC, Daggett V. Methods for molecular dynamics simulations of protein folding/unfolding in solution. Methods 2004; 34:112 - 120
- Verkhivker GM. Protein conformational transitions coupled to binding in molecular recognition of unstructured proteins: hierarchy of structural loss from all-atom Monte Carlo simulations of p27Kip1 unfolded-unbinding and structural determinants of the binding mechanism. Biopolymers 2004; 75:420 - 433
- Adzhubei AA, Sternberg JE. Left-handed polyproline II helices commonly occur in globular proteins. J Mol Biol 1993; 229:472 - 493
- Sherman SA, Greiner WH, Kirnarskiy L, Perini F, Ruddon RW. A lenear 23-residue peptide reveals a propensity to form an unusual native-like conformation. J Biomol Sci Dynamics 1995; 13:441 - 446
- Wang J, Cieplak P, Kollman PA. How well does a restrained electrostatic potential (RESP) model perform in calculating conformational energies of organicand biological molecules?. J Comput Chem 2000; 22:1048 - 1057
- Case DA, Pearlman JW, Caldwell TE, Cheatham J III, Wang WS, Ross CL, et al. AMBER 7 2002; University of California San Francisco
- Jorgensen WL, Chandrasekhar J, Madura JD, Impley RW, Klein ML. Comparison of simple potential functions for simulating liquid water. J Chem Phys 1983; 79:926 - 935
- Ryckaert JP, Ciccotti G, Berendsen HJC. Numerical intergration of the Cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J Comput Phys 1977; 23:327 - 341
- Berendsen HJC, Postma JPM, van Gunsteren WF, DiNola A, Haak JR. Molecular dynamics with coupling to an external bath. J Chem Phys 1984; 81:3684 - 3690
- Darden T, York D, Pedersen L. Particle Mesh Ewald: An N Log (N) method for Ewald sums in large systems. J Chem Phys 1993; 98:1089 - 1092
- Kabsh W, Sander C. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 1983; 22:2577 - 2637
- Kinarsky L, Nomoto M, Ikematsu Y, Hassan H, Benett EP, Cerney RL, et al. Structural analysis of the peptide substrates for mucin-type O-glycosylation. Biochem 1998; 37:12811 - 12817
- Smith LJ, Daura X, van Gunsteren WF. Assessing equilibration and convergence in biomolecular simulations. Proteins 2002; 48:487 - 496
- Massi F, Klimov D, Thirumalai D, Straub JE. Charge states rather than propensity for b-structure determine enhanced fibrillogenesis in wild-type Alzheimer's β-amyloid pep- tide compared to E22Q Dutch mutant. Protein Sci 2002; 11:1639 - 1647
- Ikebe J, Kamiya N, Ito J-I, Shindo H, Higo J. Simulation study on the disordered state of an Alzheimer's β amyloid peptide Aβ(12–36) in water consisting of random-structure, b-structural and helical clusters. Protein Sci 2007; 16:1596 - 1608
- Hutchinson EG, Thornton JM. A revised set of potentials for β-turn formation in proteins. Protein Sci 1994; 3:2207 - 2216
- Levy Y, Cho SS, Onuchic JN, Wolynes PG. A survey of flexible protein binding mechanisms and their transition states using native topology based energy landscapes. J Mol Biol 2005; 346:1121 - 1145
- Chandler D. Interfaces and the driving force of hydrophobic assembly. Nature 2005; 437:640 - 647
- Sheinerman FB, Brooks CL III. Calculations on folding of segment B1 of streptococcal protein G. J Mol Biol 1998; 278:439 - 456
- Kumar S, Nussinov R. Relationship between ion pair geometries and electrostatic strengths in proteins. Biophys J 2002; 83:1595 - 1612
- Tarus B, Straub JE, Thirumalai D. Structures and free-energy landscapes of the wild type and mutant of the Ab21-30 peptide are determined by an interplay between intrapeptide electrostatic and hydrophobic interactions. J Mol Boil 2008; 379:815 - 829
- Melchor JP, McVoy L, van Nostrand WE. Charge alteration of E22 enhence the pathogenic properties of the amyloid β-protein. J Neurochem 2000; 74:2209 - 2212
- Miravalle L, Tokuda T, Chiarle R, Giaccone G, Bugiani O, Tagliavini F, et al. Substitutions at codon 22 of Alzheimer's abeta peptide induce diverse conformational changes and apoptotic effects in human cerebral endothelial cells. J Biol Chem 2000; 275:27110 - 27116
- Van Nostrand WE, Melchor JP, Cho HS, Greenberg SM, Rebeck GW. Pathogenic effects of D23N Iowa mutant amyloid β-protein. J Biol Chem 2001; 276:32860 - 32866
- Grabovski TJ, Cho HS, Vonsattel JPG, Rebeck GW, Greenberg SM. Novel amyloid precursor protein mutation in an Iowa family with dementia and cerebral amyloid angiopathy. Ann Neurol 2001; 49:697 - 705
- Lashuel HA, Hartley DM, Petre BM, Wall JS, Simon MN, Walz T, et al. Mixture of wild-type and a pathogenic (E22G) form of Aβ40 in vitro accumulate protofibrils, including amyloid pores. J Mol Biol 2003; 332:795 - 808
- Grant MA, Lazo ND, Lomakin A, Condron MM, Arai H, Yamin G, et al. Familial Alzheimer's disease mutations alter the stability of the amyloid b-protein monomer folding nucleus. Proc Natl Acad Sci USA 2007; 104:16522 - 16527
- Chothia C, Levitte M, Richardson D. Helix to helix packing in proteins. J Mol Biol 1981; 145:215 - 250
- Efimov AV. Standard structures in proteins. Prog Biophys Mol Biol 1993; 60:201 - 239
- Efimov AV. Complementary packing of a-helices in proteins. FEBS Letters 1999; 463:3 - 6
- Harbury PB, Tidor B, Kim PS. Repacking protein cores with backbone freedom: structure prediction for coiled coil. Proc Natl Acad Sci USA 1995; 92:8408 - 8412
- Liu L, Zheng Q, Dengb Y, Cheng C-S, Kallenbach NR, Lu M. A seven-helix coiled coil. Proc Natl Acad Sci USA 2006; 103:15457 - 15462
- Zhang S, Iwata K, Lachenmann MJ, Peng JW, Li S, Stimson ER, et al. The Alzheimer's peptide Aβ adopts a collapsed coil structure in water. J Struct Biol 2000; 130:130 - 141
- Rubinstein A, Sherman S. Influence of solvent structure on the electrostatic interactions in proteins. Biophys J 2004; 87:1544 - 1557
- Rubinstein A, Shats O, Sherman S. Role of the local interactions in intrinsic conformational and secondary structure propensities of the amino acid residues in proteins. Seventh Electronic Computational Chemistry Conference (ECCC7) 2001; (http//bioinformatics.unmc.edu/ECCC7/)
- Rubinstein A, Sherman S. Lebl M, Houghten RA. The influence of the surrounding media on the structural propensities of amino acid residues in proteins. Peptides: The Wave of the Future 2001; San Diego American Peptide Society 332 - 333
- Fezoui Y, Walsh DM, Osterhout JJ. Strategies and rationales for the de novo design of a helical hairpin peptide. Proteins 1995; 4:286 - 295
- Fezoui Y, Braswell EH, Xian W, Osterhout JJ. Dissection of the de novo designed peptide αtα: stability and properties of the intact molecule and its constituent helices. Biochem 1999; 38:2796 - 2804
- Fezoui Y, Hartley DM, Walsh DM, Selkoe DJ, Osterhout JJ, Teplow DB. A de novo designed helix-turn-helix peptide forms nontoxic amyloid fibrils. Nature Struct Biol 2000; 7:1095 - 1099
- Fuxreiter M, Simon I, Friedrich P, Tompa P. Preformed structural elements feature in partner recognition by intrinsically unstructured proteins. J Mol Biol 2004; 338:1015 - 1026