Abstract
We recently solved the crystallographic structure of a dimeric form of the serpin antithrombin which has fundamentally changed the way we think about serpin polymerization. Like for other diseases that have protein deposition as a hallmark, the serpinopathies are associated with discrete inter-protomer linkage followed by subsequent association into larger fibrils and aggregates. Polymerization of the serpins is an off-pathway event that occurs during folding in the endoplasmic reticulum. Our structure reveals the nature of the polymerogenic folding intermediate, the reason that the inter-protomer linkage is hyperstable, and suggests a mechanism of lateral association of polymers into soluble fibrils and insoluble aggregates. While the basis of cellular toxicity is still unclear, novel therapeutic approaches targeting the folding intermediate or the lateral association event are now conceivable.
The serpins are serine protease inhibitors that utilize a unique and well characterized β-sheet expansion event as a necessary part of their mechanism.Citation1–Citation3 This conformational/topological change from a five-stranded (N in ) to a six-stranded β-sheet A (L in ) results in a doubling of the protein's stability, and is driven by a free-energy term of around −32 kcal/mol.Citation4 Thus, the native form of serpins is metastable in defiance of the Anfinsen principle,Citation5 requiring a folding pathway that kinetically traps the five-stranded state. This unusual protein folding requirement allows for an off-pathway event known as polymerization, where one protomer completes the A sheet of another.Citation6 For secreted serpins, this occurs in the endoplasmic reticulum (ER) where polymers are seen to accumulate as insoluble proteinaceous inclusions. Polymerization has been described for several serpins and is always associated with a loss of functional levels due to accumulation within the cell, and occasionally it is associated with the death of the secretory cells through a poorly understood mechanism.Citation7–Citation9 The best examples of the latter are provided by the Z variant of α1-antitrypsin (α1AT) leading to liver disease and the Syracuse variant of neuroserpin that causes early onset dementia.
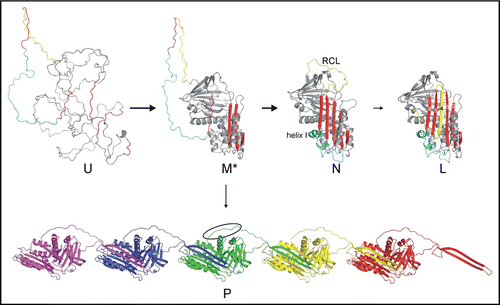
We recently solved a crystal structure of a self-terminating (closed) serpin dimer that revealed a large domain swap including the fourth and fifth strands of β-sheet A.Citation10 We then modelled an open polymerization competent polymer based on the structure (P in ) that explained their facile propagation, the hyperstability and flexibility of the inter-protomer linkage, and also suggested a structure for the polymerogenic folding intermediate (M* in ). We proposed that the final step in folding to the native state is the insertion of strand 5 into β-sheet A and the association of the coiled linker domain to the ‘bottom’ of the molecule. This event would leave the fourth strand (the reactive centre loop) accessible to serve as bait for proteolytic attack, necessary for the functioning of the serpin mechanism. While many details are yet to be confirmed, the position of certain polymerogenic mutations on and underlying strand 5A supports the proposal.Citation10
One unexpected implication of our model is the requisite unfolding and exposure of helix I and the following coiled region in linear serpin polymers. Exposure of this ‘linker region’ was verified in linear polymers of serpins antithrombin and α1AT through limited proteolysis and fluorescence studies, and explains the observation that polymers are hydrophobic and exhibit an increased propensity towards aggregation. Unglycosylated serpins typically aggregate when polymerized in vitro (with heat or low concentrations of chaotrophes), even at vanishingly low concentrations, whereas high concentrations are required to observe aggregation of glycosylated serpin polymers. We hypothesized that aggregation/precipitation occurs via lateral association of linear polymers, either through specific β-strand linkage or non-specific hydrophobic interactions involving the linker region. Sequence analysis of helix I suggest that it is a ‘frustrated’ β-strandCitation11 for several serpins including α1AT, supporting the idea that aggregates of serpin polymers form through an extended β-sheet mechanism akin to other ‘conformational diseases.’Citation12
While there are clear parallels between the ‘serpinopathies’ and conformational diseases such as Alzheimer, Huntington and the prion encephalopathies (e.g., ordered intermolecular linkage, β-sheet expansion, cell death, dementia, accumulation of insoluble aggregates, domain-swapping),Citation12,Citation13 the detailed molecular mechanism revealed by our crystal structure is unique to the serpins. Domain swaps in other proteins are generally characterized by normal activity and stability, and may not play a role in the secondary association event that leads to the toxic species.Citation14,Citation15 For serpins the domain swap leads to hyperstability and the exposure of hydrophobic regions not seen in the monomeric state. Another key difference is the manner of cellular toxicity and the nature of the toxic species. It is becoming clear that for Alzheimer, Huntington and other conformational diseases the toxic fragments are likely to be the soluble (proto)-fibrils, not the insoluble aggregates (inclusions).Citation16,Citation17 Serpin polymerization generally leads to disease through loss of secretion of the active species, and only in two special cases is it through gain-of-function cellular toxicity, and although the toxic mechanisms are incompletely resolved, they appear to involve the insoluble aggregates.
The most common cause of cirrhosis among children is the homozygous Z mutation in α1AT.Citation18 Antitrypsin is expressed at high levels by hepatocytes (1.3–3.5 g/l in blood plasma) and its expression can increase in response to infection and other stimuli.Citation19 However, only about one-third of the homozygous carriers ever manifest liver diseaseCitation18 and it never occurs in carriers of a single Z allele, indicating that hepatocytes are generally well equipped to deal with the mutant protein. Soluble Z α1AT in the ER binds to chaperones and is subsequently targeted to the ERAD pathway for clearance by the proteosome, and insoluble polymers and aggregates are thought to activate autophagy for degradation in the lysosomes.Citation20,Citation21 In such a model, accumulation of polymers and cellular toxicity only occur when the proteosomal and autophagic pathways have been saturated by the high level of expression of mutant α1AT in the liver.Citation21 In contrast, neuroserpin is expressed at low levels in neurons and mutation leads to polymerization and dementia in an autosomal dominant fashion.Citation22,Citation23 However, cell death and disease are still associated with accumulation of inclusion bodies within the ER, and the toxic mechanisms are likely to be similar.Citation9
In summary, we have elucidated a novel mechanism of serpin polymerization that involves a hyperstable domain swap of a folding intermediate. Formation of linear polymers exposes hydrophobic regions that mediate lateral polymer association and eventually leads to intra ER accretion and cellular toxicity. Our proposal suggests new avenues for the rational design of compounds to combat the diseases caused by serpin polymerization, either through targeting the folding intermediate or the lateral association of soluble polymers.
Figures and Tables
Figure 1 Serpin folding and polymerization. The pathway of serpin folding proceeds from the unfolded state (U) to the native state (N) via a stable intermediate (M*). The native conformation is the only active state, and is composed of a five-stranded A sheet (red) and a 20 residue reactive centre loop (RCL, yellow). Serpin inhibitory function requires the native conformation to be a kinetically trapped metastable state. Completion of sheet A by incorporation of the RCL as strand 4, to form the latent (L) state, results in the doubling of the serpin's thermodynamic stability (the six strands are labelled on L). Folding and unfolding of native serpins is known to proceed via a stable intermediate denoted M*, which also corresponds to the polymerogenic form.Citation24–Citation26 The key feature of the M* state is that strand 5 is not yet incorporated into sheet A, and can thus insert in an intermolecular fashion to form off-pathway polymers (P, each protomer of the pentamer is in a different colour). The polymers have complete A sheets and are thus hyperstable. As a consequence of polymerization, the linker region (cyan), containing helix I, remains unfolded. We hypothesize that the hydrophobic linker (indicated by the oval) is responsible for the lateral association of polymers into insoluble aggregates.
References
- Huntington JA, Read RJ, Carrell RW. Structure of a serpin-protease complex shows inhibition by deformation. Nature 2000; 407:923 - 926
- Gettins PG. Serpin structure, mechanism and function. Chem Rev 2002; 102:4751 - 4804
- Huntington JA. Shape-shifting serpins—advantages of a mobile mechanism. Trends Biochem Sci 2006; 31:427 - 435
- Im H, Ahn HY, Yu MH. Bypassing the kinetic trap of serpin protein folding by loop extension. Protein Sci 2000; 9:1497 - 1502
- Anfinsen CB. Principles that govern the folding of protein chains. Science 1973; 181:223 - 230
- Lomas DA, Evans DL, Finch JT, Carrell RW. The mechanism of Z alpha1-antitrypsin accumulation in the liver. Nature 1992; 357:605 - 607
- Gooptu B, Lomas DA. Polymers and inflammation: disease mechanisms of the serpinopathies. J Exp Med 2008; 205:1529 - 1534
- Perlmutter DH, Brodsky JL, Balistreri WF, Trapnell BC. Molecular pathogenesis of alpha-1-antitrypsin deficiency-associated liver disease: a meeting review. Hepatology 2007; 45:1313 - 1323
- Davies MJ, Lomas DA. The molecular aetiology of the serpinopathies. Int J Biochem Cell Biol 2008; 40:1273 - 1286
- Yamasaki M, Li W, Johnson DJ, Huntington JA. Crystal structure of a stable dimer reveals the molecular basis of serpin polymerization. Nature 2008; 455:1255 - 1258
- Kallberg Y, Gustafsson M, Persson B, Thyberg J, Johansson J. Prediction of amyloid fibril-forming proteins. J Biol Chem 2001; 276:12945 - 12950
- Carrell RW, Lomas DA. Conformational disease. Lancet 1997; 350:134 - 138
- Lomas DA, Carrell RW. Serpinopathies and the conformational dementias. Nat Rev Genet 2002; 3:759 - 768
- Bennett MJ, Sawaya MR, Eisenberg D. Deposition diseases and 3D domain swapping. Structure 2006; 14:811 - 824
- Liu Y, Eisenberg D. 3D domain swapping: as domains continue to swap. Protein Sci 2002; 11:1285 - 1299
- Ferreira ST, Vieira MN, De Felice FG. Soluble protein oligomers as emerging toxins in Alzheimer's and other amyloid diseases. IUBMB Life 2007; 59:332 - 345
- Stefani M, Dobson CM. Protein aggregation and aggregate toxicity: new insights into protein folding, misfolding diseases and biological evolution. J Mol Med 2003; 81:678 - 699
- Fairbanks KD, Tavill AS. Liver disease in alpha1-antitrypsin deficiency: a review. Am J Gastroenterol 2008; 103:2136 - 2141
- Perlmutter DH. Pathogenesis of chronic liver injury and hepatocellular carcinoma in alpha-1-antitrypsin deficiency. Pediatr Res 2006; 60:233 - 238
- Schmidt BZ, Perlmutter DH. Grp78, Grp94 and Grp170 interact with alpha1-antitrypsin mutants that are retained in the endoplasmic reticulum. Am J Physiol Gastrointest Liver Physiol 2005; 289:444 - 455
- Perlmutter DH. Autophagic disposal of the aggregation-prone protein that causes liver inflammation and carcinogenesis in alpha-1-antitrypsin deficiency. Cell Death Differ 2009; 16:39 - 45
- Galliciotti G, Sonderegger P. Neuroserpin. Front Biosci 2006; 11:33 - 45
- Miranda E, Lomas DA. Neuroserpin: a serpin to think about. Cell Mol Life Sci 2006; 63:709 - 722
- Kim D, Yu MH. Folding pathway of human alpha1-antitrypsin: characterization of an intermediate that is active but prone to aggregation. Biochem Biophys Res Commun 1996; 226:378 - 384
- Devlin GL, Chow MK, Howlett GJ, Bottomley SP. Acid Denaturation of alpha(1)-Antitrypsin: Characterization of a Novel Mechanism of Serpin Polymerization. J Mol Biol 2002; 324:859 - 870
- Tew DJ, Bottomley SP. Probing the equilibrium denaturation of the serpin alpha(1)-antitrypsin with single tryptophan mutants; evidence for structure in the urea unfolded state. J Mol Biol 2001; 313:1161 - 1169