Abstract
The human PrP gene (PRNP) has two major polymorphic codons: 129 for methionine (M) or valine (V), and 219 for glutamate (E) or lysine (K). The PRNP heterozygotes appear to be protected from sporadic CJD compared to the PRNP homozygotes. The molecular mechanism responsible for these protective effects of PRNP heterozygosity has remained elusive. In this review, we describe the inhibition of PrP conversion observed in a series of transmission studies using PRNP heterozygous animal models. In vCJD infection, the conversion incompetent human PrP 129V molecules showed an inhibitory effect on the conversion of human PrP 129M molecules in the 129M/V heterozygous mice. Furthermore, though the human PrP 219E and PrP 219K were both conversion competent in vCJD infection, these conversion competent PrP molecules showed an inhibitory effect in the 219E/K heterozygous animals. To explain this heterozygous inhibition, we propose a possible mechanism designated as the stone fence model.
Introduction
Creutzfeldt-Jakob disease (CJD), scrapie, and bovine spongiform encephalopathy are lethal transmissible neurodegenerative diseases caused by an abnormal isoform of prion protein (PrPSc) that is converted from the normal cellular isoform (PrPC).Citation1 The human PrP gene (PRNP) has two major polymorphic codons: 129 for methionine (M) or valine (V), and 219 for glutamate (E) or lysine (K).Citation2,Citation3 These PRNP polymorphisms affect the susceptibility to sporadic (sCJD), variant (vCJD), or iatrogenic CJD.Citation4–Citation8 In particular, the PRNP heterozygotes appear to be protected from sCJD compared to the PRNP homozygotes. The frequency of the PRNP 129M/V genotype in sCJD is significantly lower than that in the normal population.Citation5 Moreover, the PRNP 219E/K genotype is absent in sCJD patients.Citation9 The molecular mechanism responsible for these protective effects of PRNP heterozygosity has remained elusive.
In this review, we describe the inhibition of PrP conversion observed in a series of transmission studies using PRNP heterozygous animal models.Citation10,Citation11 To explain this heterozygous inhibition, we propose a possible mechanism designated as the stone fence model.
Two Modes of Heterozygous Inhibition
Inhibition by the conversion incompetent PrP molecules.
vCJD prions (genotype: 129M/M and 219E/E) can be transmitted to knock-in mice expressing human PrP with 129M/M (Ki-Hu129M/M) or with 129M/V (Ki-Hu129M/V), but not to those with 129V/V (Ki-Hu129V/V).Citation10 In transmission experiments using vCJD prions, we found an inhibitory effect of the conversion incompetent PrP 129V molecules in the 129M/V heterozygous animals. The amount of PrPSc in the spleens of Ki-Hu129M/V mice intraperitoneally inoculated with vCJD prions was much lower than that in the spleens of Ki-Hu129M/M mice ().Citation11 Moreover, the amount of PrPSc in Ki-Hu129M/V mice was even lower than that in hemizygous knock-in mice expressing human PrP 129M from one allele (Ki-Hu129M/0), which express half the level of PrP 129M compared with Ki-Hu129M/M mice. Thus, we confirmed that the decreased PrPSc accumulation in Ki-Hu129M/V mice was not due only to the expression level of PrP 129M. These findings clearly showed that the conversion incompetent PrP 129V molecules exerted an inhibitory effect on the conversion of PrP 129M molecules in the 129M/V heterozygous animals.
Previous studies have demonstrated that the conversion incompetent PrP molecules exhibit inhibitory effects on the conversion of the co-existing conversion competent PrP. This type of inhibition has been referred to as a dominant negative effect. When the endogenous mouse PrP gene was ablated, transgenic mice expressing exogenous human PrP or hamster PrP became more susceptible to human prions or hamster prions, respectively.Citation12,Citation13 In a cell-free conversion system using mouse and hamster PrP, the conversion but not the binding to PrPSc was inhibited by the conversion incompetent PrP in a dosedependent manner.Citation14 Furthermore, dominant negative mutations in mouse PrP have been studied intensively due to their potential for therapeutic applications.Citation15–Citation22 In accord with these reports, the conversion incompetent human PrP 129V molecules in our study showed an inhibitory effect on the conversion of the human PrP 129M molecules.
Inhibition by the conversion competent PrP molecules.
When we performed intraperitoneal inoculation of vCJD prions into knock-in mice expressing human PrP with 129M/M and 219E/E (Ki-Hu219E/E, a synonym of Ki-Hu129M/M), 129M/M and 219K/K (Ki-Hu219K/K), or 129M/M and 219E/K (Ki-Hu219E/K), we made two important findings. (1) Ki-Hu219K/K mice showed high susceptibility to vCJD prions. (2) Nevertheless, the heterozygous Ki-Hu219E/K mice showed the lowest susceptibility among the knock-in mice with polymorphism at codon 219.Citation11 The amount of PrPSc and the number of PrP-positive follicular dendritic cells in the spleens of Ki-Hu219K/K mice were higher than those of Ki-Hu219E/E mice (). By contrast, the amount of PrPSc accumulation in Ki-Hu219E/K mice was even lower than that in the hemizygous Ki-Hu219E/0 mice or Ki-Hu219K/0 mice. Thus, though the human PrP 219E and PrP 219K were both conversion competent in vCJD infection, these conversion competent PrP molecules showed an inhibitory effect in the 219E/K heterozygous animals.
The Stone Fence Model of Heterozygous Inhibition
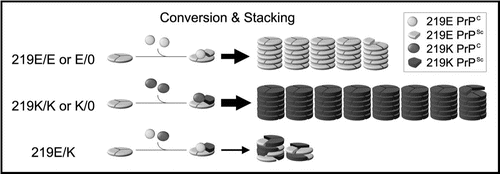
The molecular mechanisms responsible for the inhibitory effect of the conversion incompetent PrP have been studied previously.Citation14,Citation23,Citation24 Briefly, though the conversion incompetent PrP molecules are not converted into PrPSc, they bind to PrPSc and are incorporated into amyloid fibrils with the conversion competent PrP molecules. Based on these previous findings, we propose a possible mechanism to explain the inhibitory effect of the conversion competent PrP as well as the conversion incompetent PrP. We designated the mechanism as the stone fence model ().
PrPC is converted to PrPSc and then piled up into amyloid fibrils according to the nucleated polymerization hypothesis.Citation25 In homozygous (219E/E or 219K/K) or hemizygous (219E/0 or K/0) animals, the conversion results in a single PrPSc population (). This means that the same blocks (the same PrPSc) would be piled up into the amyloid fibrils without any delay. By contrast, in the heterozygous 219E/K animals, the conversion results in at least two distinct PrPSc populations (219E PrPSc or 219K PrPSc) with different structures. These two PrPSc blocks would be piled up into the same fibril just like a stone fence composed of heterologous blocks. However, the fibril elongation would be delayed because the two types of PrPSc blocks interfere with each other due to their incompatible structures. Thus, the two PrPSc populations with different structures can act as decelerators of each other in the process of stacking.
The decelerator hypothesis based on the stone fence model is compatible with the possible mechanism for the inhibitory effect of the conversion incompetent PrP molecules.Citation14,Citation23 The conversion incompetent PrP is incorporated into amyloid fibrils with the conversion competent PrP, but acts as a decelerator at the conversion step. Therefore, the heterozygous inhibition can be caused both by the conversion incompetent PrP and the conversion competent PrP, and can occur both at the conversion step and the stacking step. Since it remains to be determined whether the human PrP 219E is efficiently converted by 219K PrPSc, we cannot rule out the possibility that the inhibition in the 219E/K heterozygous animals also occurs at the conversion step in addition to the stacking step.
Mysterious Phenomena in Prion Diseases Revisited With The Stone Fence Model
The stone fence model can explain the prion strain interference.Citation29,Citation30 The preceding infection with a prion strain prior to the superinfection with another prion strain interferes with the replication of the superinfected strain.Citation31 According to the stone fence model (), the pre-existing PrPSc and the superinfected PrPSc might be piled up into the same fibril, but act as decelerators of each other due to their incompatible structures. To pile up the two distinct PrPSc populations into the same fibril, still unidentified interactions between the PrPSc molecules such as inter-oligomer interactionCitation32 or protofibril stackingCitation33 might underlie the stacking step. Thus, if the pre-existing PrPSc dominates enough, the elongation of the amyloid fibrils seeded by the superinfected PrPSc is decelerated by the pre-existing PrPSc. Meanwhile, the efficacy of interference depends on the combination of the prion strains co-infected. Scrapie 22L strain but not Chandler strain interfered with the replication of the superinfected Gerstmann-Sträussler-Scheinker disease Fukuoka-1 strain.Citation34 The compatibility between the two types of PrPSc blocks might determine the efficacy of interference.
The stone fence model can also account for the absence of the PRNP 219E/K genotype in sCJD patients.Citation9 In our experiments using vCJD prions, the human PrP 219E and PrP 219K were both conversion competent, whereas the susceptibility of the 219E/K heterozygous animals was even lower than that of the hemizygous animals. Furthermore, intracerebral transmission experiments using sCJD prions revealed that the human PrP 219E and PrP 219K were both conversion competent also in sCJD infection.Citation11,Citation35 Though the susceptibility of the 219E/K heterozygous animals to sCJD prions remains to be determined, the absence of the PRNP 219E/K genotype in sCJD patients is probably due to the heterozygous inhibition. Even though the human PrP 219E or PrP 219K spontaneously converts into PrPSc, subsequent fibril formation and elongation would be decelerated because the heterologous PrPSc blocks act as decelerators of each other.
Lessons From the Heterozygous Inhibition Experiments
Mouse PrP 218K (corresponding to human PrP 219K) molecules are conversion incompetent in mouse scrapie infection and show dominant negative effects both in vitro and in vivo.Citation15–Citation20 Therefore, the absence of the PRNP 219E/K genotype in sCJD patients was formerly explained by the dominant negative effect. However, our study revealed that the human PrP 219K molecules are conversion competent in sCJD infection as well as vCJD infection. This discrepancy suggested that the mutations in mouse PrP exhibit different effects from those of the corresponding mutations in human PrP as regards the conversion competence. This could have great significance for transgenic models expressing mouse PrP with mutations corresponding to the human pathogenic mutations. Otherwise, the distinct prion strains used in the experiments might underlie the discrepancy.Citation19
To compare precisely the susceptibility of the experimental animals with different PrP genotypes, knock-in mice including heterozygous mice have an advantage over transgenic mice because they have identical genetic backgrounds, identical PrP expression levels, and equivalent expression from the heterozygous genes.Citation10 Furthermore, the hemizygous knock-in mice express exactly half the level of PrP compared to the homozygous mice.Citation11 Since the expression level of PrP affects the length of the incubation period regardless of the PrP genotype, the heterozygous and the hemizygous knock-in mice are both indispensable to analyze the heterozygous inhibition.
Conclusion
The present study, together with evidence from other groups, suggests that heterozygous inhibition is a universal phenomenon that can be caused by both conversion incompetent PrP and conversion competent PrP, or by both PrPC-heterozygosity and PrPSc-heterozygosity. The decelerator hypothesis based on the stone fence model paves the way for the solution of this phenomenon. To determine whether the efficacy of heterozygous inhibition is affected by the infected prion strain or the host PrP genotype, other heterozygous models need to be examined.
Abbreviations
| PrP | = | prion protein |
| PrPC | = | normal cellular isoform of PrP |
| PrPSc | = | abnormal isoform of PrP |
| CJD | = | Creutzfeldt-Jakob disease |
| vCJD | = | variant CJD |
| sCJD | = | sporadic CJD |
| PRNP | = | human PrP gene |
Figures and Tables
Figure 1 Heterozygous inhibition in vCJD infection. Western blot analysis of PrPSc in the spleens of knock-in mice intraperitoneally inoculated with vCJD prions. The amount of PrPSc in the 129M/V heterozygous mice was even lower than that in the 129M/0 hemizygous mice. Furthermore, the amount of PrPSc was the highest in the 219K/K mice, whereas the PrPSc accumulation in the 219E/K heterozygous mice was even lower than that in the 219E/0 hemizygous mice or 219K/0 hemizygous mice. Therefore, we found that both the conversion incompetent PrP and the conversion competent PrP showed inhibitory effects in the heterozygous animals.
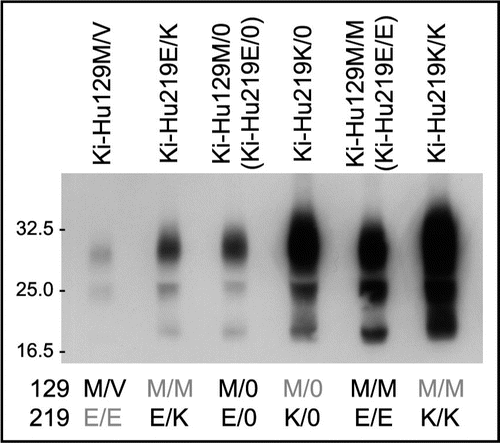
Figure 2 The stone fence model: a possible mechanism of the heterozygous inhibition in vCJD infection. PrPC is converted to PrPSc and then piled up into amyloid fibrils according to the nucleated polymerization hypothesis and the trimeric models.Citation25–Citation28 In the homozygous (219E/E or K/K) or the hemizygous (219E/0 or K/0) animals, the PrPSc blocks are piled up into the amyloid fibrils without delay because only a uniform PrPSc population exists. Though the initial seed is 219E PrPSc also in the 219K/K or 219K/0 animals, the resulting 219K PrPSc acts as a new seed in the subsequent steps and are efficiently piled up. Therefore, the inhibitory effect of the initial 219E PrPSc seed is negligible in these animals. By contrast, in the heterozygous (219E/K) animals, at least two PrPSc populations are generated. These two PrPSc blocks are piled up into the same fibril just like a stone fence composed of heterologous blocks. The two types of PrPSc blocks interfere with each other due to their incompatible structures and delay the fibril elongation. Thus, the distinct PrPSc populations act as decelerators of each other in the heterozygous animals.
Acknowledgements
We thank H. Kudo and Y. Ishikawa for technical assistance, and B. Bell for critical review of the manuscript. This study was supported by the Promotion of Fundamental Studies in Health Science of National Institute of Biomedical Innovation (S.M., and T.K.), a grant from the Ministry of Health, Labor, and Welfare (A.K., K.T., S.M., and T.K.), and a Grant-in Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology (A.K., K.T. and T.K.).
References
- Prusiner SB, Scott MR, DeArmond SJ, Cohen FE. Prion protein biology. Cell 1998; 93:337 - 348
- Doh-ura K, Tateishi J, Sasaki H, Kitamoto T, Sakaki Y. Pro-Leu change at position 102 of prion protein is the most common but not the sole mutation related to Gerstmann-Sträussler syndrome. Biochem Biophys Res Commun 1989; 163:974 - 979
- Kitamoto T, Tateishi J. Human prion diseases with variant prion protein. Phil Trans R Soc Lond B Biol Sci 1994; 343:391 - 398
- Collinge J, Palmer MS, Dryden AJ. Genetic predisposition to iatrogenic Creutzfeldt-Jakob disease. Lancet 1991; 337:1441 - 1442
- Palmer MS, Dryden AJ, Hughes JT, Collinge J. Homozygous prion protein genotype predisposes to sporadic Creutzfeldt-Jakob disease. Nature 1991; 352:340 - 342
- Doh-ura K, Kitamoto T, Sakaki Y, Tateishi J. CJD discrepancy. Nature 1991; 353:801 - 802
- Zeidler M, Stewart G, Cousens SN, Estibeiro K, Will RG. Codon 129 genotype and new variant CJD. Lancet 1997; 350:668
- Alperovitch A, Zerr I, Pocchiari M, Mitrova E, de Pedro Cuesta J, Hegyi I, et al. Codon 129 prion protein genotype and sporadic Creutzfeldt-Jakob disease. Lancet 1999; 353:1673 - 1674
- Shibuya S, Higuchi J, Shin R-W, Tateishi J, Kitamoto T. Codon 219 Lys allele of PRNP is not found in sporadic Creutzfeldt-Jakob disease. Ann Neurol 1998; 43:826 - 828
- Asano M, Mohri S, Ironside JW, Ito M, Tamaoki N, Kitamoto T. vCJD prion acquires altered virulence through trans-species infection. Biochem Biophys Res Commun 2006; 342:293 - 299
- Hizume M, Kobayashi A, Teruya K, Ohashi H, Ironside JW, Mohri S, et al. Human prion protein (PrP) 219K is converted to PrPSc but shows heterozygous inhibition in vCJD infection. J Biol Chem 2009; 284:3603 - 3609
- Telling GC, Scott M, Mastrianni J, Gabizon R, Torchia M, Cohen FE, et al. Prion propagation in mice expressing human and chimeric PrP transgenes implicates the interaction of cellular PrP with another protein. Cell 1995; 83:79 - 90
- Büeler H, Aguzzi A, Sailer A, Greiner RA, Autenried P, Aguet M, et al. Mice devoid of PrP are resistant to scrapie. Cell 1993; 73:1339 - 1347
- Horiuchi M, Priola SA, Chabry J, Caughey B. Interactions between heterologous forms of prion protein: binding, inhibition of conversion, and species barriers. Proc Natl Acad Sci USA 2000; 97:5836 - 5841
- Kaneko K, Zulianello L, Scott M, Cooper CM, Wallace AC, James TL, et al. Evidence for protein X binding to a discontinuous epitope on the cellular prion protein during scrapie prion propagation. Proc Natl Acad Sci USA 1997; 94:10069 - 10074
- Perrier V, Kaneko K, Safar J, Vergara J, Tremblay P, DeArmond SJ, et al. Dominant-negative inhibition of prion replication in transgenic mice. Proc Natl Acad Sci USA 2002; 99:13079 - 13084
- Kishida H, Sakasegawa Y, Watanabe K, Yamakawa Y, Nishijima M, Kuroiwa Y, et al. Non-glycosylphosphatidylinositol (GPI)-anchored recombinant prion protein with dominant-negative mutation inhibits PrPSc replication in vitro. Amyloid 2003; 11:14 - 20
- Crozet C, Lin Y-L, Mettling C, Mourton-Gilles C, Corbeau P, Lehmann S, et al. Inhibition of PrPSc formation by lentiviral gene transfer of PrP containing dominant negative mutations. J Cell Sci 2004; 117:5591 - 5597
- Atarashi R, Sim VL, Nishida N, Caughey B, Katamine S. Prion strain-dependent differences in conversion of mutant prion proteins in cell culture. J Virol 2006; 80:7854 - 7862
- Furuya K, Kawahara N, Yamakawa Y, Kishida H, Hachiya NS, Nishijima M, et al. Intracerebroventricular delivery of dominant negative prion protein in a mouse model of iatrogenic Creutzfeldt-Jakob disease after dura graft transplantation. Neurosci Lett 2006; 402:222 - 226
- Toupet K, Compan V, Crozet C, Mourton-Gilles C, Mestre-Francés N, Ibos F, et al. Effective gene therapy in a mouse model of prion diseases. PLoS One 2008; 23:e2773
- Ott D, Taraborrelli C, Aguzzi A. Novel dominant-negative prion protein mutants identified from a randomized library. Protein Eng Des Sel 2008; 21:623 - 629
- Meier P, Genoud N, Prinz M, Maissen M, Rülicke T, Zurbriggen A, et al. Soluble dimeric prion protien binds PrPSc in vivo and antagonizes prion disease. Cell 2003; 113:49 - 60
- Lee CI, Yang Q, Perrier V, Baskakov IV. The dominant-negative effect of the Q218K variant of the prion protein does not require protein X. Protein Sci 2007; 16:2166 - 2173
- Harper JD, Lansbury PT Jr. Models of amyloid seeding in Alzheimer's disease and scrapie: mechanistic truths and physiological consequences of the time-dependent solubility of amyloid proteins. Annu Rev Biochem 1997; 66:385 - 407
- Govaerts C, Wille H, Prusiner SB, Cohen FE. Evidence for assembly of prions with left-handed β-helices into trimers. Proc Natl Acad Sci USA 2004; 101:8342 - 8347
- DeMarco ML, Daggett V. From conversion to aggregation: Protofibril formation of the prion protein. Proc Natl Acad Sci USA 2004; 101:2293 - 2298
- Langedijk JPM, Fuentes G, Boshuizen R, Bonvin AMJJ. Two-rung model of a left-handed β-helix for prions explains species barrier and strain variation in transmissible spongiform encephalopathies. J Mol Biol 2006; 360:907 - 920
- Dickinson AG, Fraser H, Meikle VM, Outram GW. Competition between different scrapie agents in mice. Nat New Biol 1972; 237:244 - 245
- Manuelidis L. Vaccination with an attenuated Creutzfeldt-Jakob disease strain prevents expression of a virulent agent. Proc Natl Acad Sci 1998; 95:2520 - 2525
- Bartz JC, Kramer ML, Sheehan MH, Hutter JAL, Ayers JI, Bessen RA, et al. Prion interference is due to a reduction in strain-specific PrPSc levels. J Virol 2007; 81:689 - 697
- Gerber R, Voitchovsky K, Mitchel C, Tahiri-Alaoui A, Ryan JF, Hore PJ, et al. Interoligomer interactions of the human prion protein are modulated by the polymorphism at codon 129. J Mol Biol 2008; 381:212 - 220
- Arimon M, Díez-Pérez I, Kogan MJ, Durany N, Giralt E, Sanz F, et al. Fine structure study of Aβ1-42 fibrillogenesis with atomic force microscopy. FASEB J 2005; 19:1344 - 1346
- Nishida N, Katamine S, Manuelidis L. Reciprocal interference between specific CJD and scrapie agents in neural cell cultures. Science 2005; 310:493 - 496
- Kobayashi A, Asano M, Mohri S, Kitamoto T. Cross-sequence transmission of sporadic Creutzfeldt-Jakob disease creates a new prion strain. J Biol Chem 2007; 282:30022 - 30028