Abstract
The complete molecular characterization of human genetic prion diseases from different backgrounds is important for clinical diagnosis and epidemiological classification. The characterization of the PRNP gene should always include the description of the pathogenic mutation, as well as the status at each allele of the polymorphic codon 129 (M129V), a well-established susceptibility marker and phenotypic variability factor for different types of human prion diseases. Indeed, the phenotypical expression of two of the most common mutations in the human PRNP gene associated with genetic prion diseases, D178N and E200K, is clearly modulated by the codon 129 polymorphism. Here, we describe two simple, fast, cost-effective and suited for high-throughput protocols to resolve cis-trans ambiguities between these mutations respect the M129V polymorphism. This methodology is based on differential amplification by allele-specific primers using Real Time PCR monitored by SYBR® Green dye. The main advantages of these protocols are their relative simplicity and the reduced cost compared to other methods such as cloning protocols, and that it may be readily applicable to the characterization of other mutations with codon 129-dependent expression, e.g. P102L.
Introduction
Prion diseases are a group of neurodegenerative diseases that have in common a key role for the prion protein in their pathogenesis.Citation1 Clinically, these diseases exhibit rapidly progressive symptoms of cognitive and motor dysfunctions, and neuropathologically are characterized by neuronal loss, gliosis, spongiosis and accumulation of aggregates of PrPSc, a misfolded and beta-sheet-rich isoform of a normal cellular protein, PrPC.Citation2 From an etiological standpoint, prion diseases can be classified as sporadic (idiopathic), acquired, or genetic cases. Sporadic Creutzfeldt-Jakob disease (sCJD) is the most frequent human prion disease (accounting for ∼85% of total) and have a worldwide distribution with a roughly even incidence of about 1.0 cases per million per year. Acquired human prion diseases are rare and include iatrogenic CJD, kuru and variant CJD.Citation3 Genetic human prion diseases represent about 10–15% of cases and are associated with one of the more than 30 recognized individual coding mutations in PRNP gene on chromosome 20p12.Citation4 Kindreds with genetic prion diseases have been described with phenotypes of classical CJD (gCJD), Gerstmann-Straüssler-Sheinker syndrome (GSS) or fatal familial insomnia (FFI) associated to specific mutations.Citation4,Citation5 However, the recent widespread availability of genetic testing has allowed diagnosis of genetic prion disease in patients with atypical clinical and neuropathological features that do not fit with this clinicopathological-oriented classification. Moreover, the neuropathological significant overlapping of this atypical cases with other neurodegenerative conditions such as Alzheimer disease, Lewy body dementia or frontal lobe degeneration points out to the relevance of genetic analysis for definitive diagnosis and the correct classification of inherited forms according to pathogenic mutations.
On the other hand, the polymorphic codon 129 of the PRNP gene is a well-established susceptibility marker, as well as a phenotype modulator for different types of human prion diseases.Citation4,Citation6 One of the most common mutations in the human PRNP gene codes for a change of asparagine instead of aspartic acid at position 178 (D178N), and represents a truly remarkable example of the influence of codon 129 on the phenotypic expression of the disease. According to the classical clinicopathological classification of genetic prion diseases, this mutation is shared by two phenotypic distinct entities: gCJD and FFI.Citation6 In general, it is accepted that when the mutation occurs in a M129 allele (M129-N178) the clinicopathological manifestations are those characteristic of FFI. On the contrary, when the mutation occurs in a Val129 allele (V129-N178) the clinicopathological manifestations are related to gCJD. Homocigosity at codon 129 is associated with more severe phenotypes.
However, recently it has been reported that this genotype-phenotype association is not absolute, since several patients have been identified with a gCJD phenotype and M129-D178N genotype, suggesting that FFI and gCJD represent two extreme manifestations of a continuous disease spectrum.Citation7
Similarly, other common gCJD subtype associated with a mutation at codon 200 (E200K) shows a phenotypic expression associated with the polymorphism at codon 129, where homozygous genotypes (M129M or V129V) present clinical manifestations more severe than the heterozygous genotype (M129V).Citation8–Citation10
Recent descriptions have favored the use of a genetic classification rather than the traditional clinicopathological one. Additionally to the description of the mutation, this molecular genetic classification should always include information on the status of codon 129 at cis (same allele) or trans (alternate allele) positions, which is very relevant for the correct diagnosis of the disease and epidemiological classification.Citation11,Citation12
Here, we describe two simple, fast and reproducible methods to resolve cis-trans ambiguities between mutated alleles for the M129V-D178N and M129V-E200K genetic prion disease backgrounds based on the use specific primers and real-time PCR monitored by SYBR® green dye.
Results and Discussion
In order to resolve cis-trans ambiguities between mutated alleles for the M129V-D178N and M129V-E200K genetic backgrounds, we have designed two sets of specific amplification reactions based on the use specific primers and real-time PCR protocols monitored by the SYBR® green dye. These protocols are based on the differential amplification efficiency for each allele with four different sets of amplication primers pairs, depending on whether they present a perfect match with both primers (located at the polymorphic codon 129 and on the mutated codon 200K or 178N, respectively).
As estimates for the efficiency of the amplification reactions, we used the cycle threshold (Ct) parameter, defined as the number of amplification cycles required for the fluorescent signal to cross a threshold. This threshold value is fixed at the exponential phase of the amplification cycles. Thus, low Ct values are indicative of specific amplification reactions, whereas higher Ct values are expected for non-specific amplifications.
By this method and using the experimental conditions described above in a StepOne real-time PCR System or a 7500 real-time PCR Systems, we were capable to resolve cis-trans ambiguities between mutated alleles for the M129V-D178N and M129V-E200K genetic prion disease backgrounds. The PRNP genotypes were determined by differential amplification in the two different sets of four reaction mixtures, depending on the mutation to be studied. As shown in the , positive (or specific) amplifications resulted in Ct values below 25 cycles, whereas negative (or non-specific) amplifications have Ct values above 25 cycles. Negative controls did not show amplification before cycle 35. The difference in the Ct values between positive and negative reactions was always of at least four cycles, which indicates that the specific amplifications are more than 16-fold more efficient compared to the non-specific ones.
shows the amplification plots in a StepOne real-time PCR System of the different PRNP genotypes characterized, namely: (1) (M129-N178) + (V129-D178) (Plot A) suggesting a IFF diagnosis, (2) (M129-D178) + (V129-N178) (Plot B) suggesting a gCJD diagnosis. Similarly for the mutation E200K, (M129-K200) + (V129-E200) (Plot C) and (M129-E200) + (V129-K200) (Plot D), which are indicative of gCJD diagnosis with different phenotypes.
The accuracy of the genotyping and allelic discrimination for all patients analysed in this work by the real-time amplification protocols was corroborated by cloning and sequence analysis with a perfect concordance in every case. In order to explore the versatility and robustness of these genotyping protocols, we studied their applicability to lower amounts of DNA material (from 50 ng down to 1.28 ng per well). With decreasing amounts of DNA, we observed that the ΔCt between positive and negative reactions were similar or smaller, but always sufficient to differentiate them ( and ). In our hands, the use of lower amount of DNA (e.g., 0.5 ng per well) yielded unreliable results.
Two methods have been commonly used for allelic discrimination of the PRNP gene, namely PCR-RFLP (PCR-Restriction Fragment Length Polymorphism) analysis and molecular cloning in E. coli. PCR-RFLP analysis by mean of simultaneous digestion of the complete coding region of PRNP with MaeII and Tth111I (for D178N) or MaeII and BsmAI (E200K) is an inexpensive, buterror-prone method, due to possible incomplete restriction enzyme digestion. Cloning in E. coli of the amplified PRPN gene, followed by the characterization by sequence analysis of several independent clones is a very reliable, although tedious and time-consuming method. The screening for point mutations performed by SSCP (single-strand conformational polymorphism) analysis, as well as the use of FRET (Fluorescent Resonance Energy Transfer) and TaqMan® probes in Real-time PCR are valuable but costly methods, and they may require further processing by complementary methods to determine the exact nature of the changes detected and to determine the allelic phase.
In conclusion, the methods described here for the allelic discrimination of D178N and E200K mutation carriers and the polymorphic codon 129 are accurate, simple, fast (2 h approximately), cost-effective and scalable to high-throughput systems, and likely easily transportable to other mutations that may show dependence of the polymorphism at codon 129, such as the P102L mutation.Citation14,Citation15
Thus, we proposed the use of these methods for complete characterization of the different genetic prion diseases backgrounds for clinical diagnosis and epidemiological classification.
Materials and Methods
Human genomic DNA isolation and sequence analysis.
Genomic DNA was extracted from human peripheral whole blood by NucleoSpin® Blood L kit (Macherey-Nagel) according to the manufacturer's instructions. The samples analyzed included DNA from patients with suspected prion diseases whose samples were submitted to our laboratory for diagnostic purposes. For all cases, the coding region of PRNP gene was amplified by PCR and the amplicons analyzed by DNA sequencing following standard procedures. Briefly, the amplification reaction was carried out with 50 ng of genomic DNA and 1 unit of Taq DNA Polymerase (Applied Biosystems) in a volume of 50 µl. The final concentrations of other reactants were: 1× Taq DNA Polymerase Buffer, 0.1 mM dNTPs, 1.5 mM MgCl2 and 0.1 µM of each primer (forward primer 5′-TGC TTT TGT CCT AAG TGC TTC AGA G-3′ and reverse primer 5′-GGG GCT TGA CCA GCA TCT CA-3′). The PCR cycling conditions were as follows: initial denaturation at 96°C for 3 min followed by 35 cycles of 96°C for 30 s, 58°C for 30 s and 72°C for 1 min and a final extension at 72°C for 10 min. Amplification was performed in a Mastercycler epGradient S thermal cycler (Eppendorf).
An aliquot of 2 µl of the product was used in sequencing reactions (BigDye® Terminator v1.1 Cycle Sequencing Kit, Applied Biosystems), using the above primers at the same final concentration. The amplified fragments were sequenced using an ABI PRISM® 377 Analyzer (Applied Biosystems) and analyzed by Chromas v2.33 software.
For this study, we selected cases with the D178N (4 cases) or E200K (11 cases) mutations, heterozygous M/V at codon 129.
Oligonucleotide primers design.
Primers were designed based on PRNP reference sequence (NCBI Reference Sequence: NG_009087.1, Homo sapiens prion protein (PrP) on chromosome 20) to match the ATG codon coding for 129M (Primer 1) or the GTG codon coding for 129V (Primer 2) for the forward primers. The reverse primers were designed to match either: (1) the wildtype codon GAC coding for 178D (Primer 3) or the mutated codon AAC coding for 178N (Primer 4), or (2) the wildtype codon GAG coding for 200E (Primer 5) or the mutated codon AAG coding for 200K (Primer 6) (). Their 3'-end nucleotides constitute the polymorphic or mutated site.
DNA amplification by real-time PCR.
For the determination of the segregation of polymorphism 129 and mutations at codon 178 or codon 200, the primers were combined in two different sets of four reaction mixtures, depending on the mutation to be studied ().
Each PCR reaction mixture contained the following reagents in a final volume of 10 µl: 1× Power SYBR® green PCR Master Mix (Applied Biosystems), 0.3 µM of each primer and 50 ng of genomic DNA. All the reactions were run in duplicate, and the same reaction mixtures without genomic DNA were used as negative controls. The PCR amplification protocol for all the reactions was as follows: initial AmpliTaq Gold DNA Polymerase activation at 95°C for 10 min and 40 cycles with denaturation at 95°C for 15 s and annealing + extension at 66°C for 1 min. Amplification was performed and monitored in a StepOne real-time PCR System (48-well format) or in a 7500 Real-time PCR System (96-well format) (Applied Biosystems). The lengths of the expected amplification products are shown in .
DNA amplification and PRNP fragment cloning.
In order to corroborate the results from the allele discrimination by real-time PCR, the coding region of PRNP gene containing the codon for E200K or the D178N mutations and the codon 129 was amplified and the individual alleles cloned in E. coli following standards procedures.Citation13 In brief, the PCR amplification reaction was carried out with 250 ng of genomic DNA and 1 unit of Pfu Ultra DNA Polymerase (Stratagene) in a volume of 50 µl. The final concentrations of other constituents were: 1× Pfu Ultra DNA Polymerase Buffer, 0.1 mM dNTPs, 1.5 mM MgCl2 and 0.1 µM of each primer (forward primer 5′-ATG CTA CGC TAG GAT CCG ACC TTC CTC ATC CCA CTA TCA GGA AGA TG-3′ and reverse primer 5′-ATC GCA TCC GAA GCT TGC AGT CAT TAT GGC GAA CC-3′). These primers include the restriction sites for BamHI and HindIII, respectively. The PCR cycling conditions were as follows: initial denaturation at 96°C for 3 min followed by five cycles of 94°C for 30 s, 55°C for 30 s and 72°C for 45 s; 30 cycles of 94°C for 30 s, 60°C for 30 s and 72°C for 45 s, and a final extension at 72°C for 7 min. Amplification was performed in a Mastercycler epGradient S thermal cycler (Eppendorf). An amplicon of 754 bp was expected.
The plasmid pcDNA 3.1 (Invitrogen) and the amplicon from above PCR reaction were digested with BamHI and HindIII restriction enzymes (Roche), and lately and ligated by T4 DNA ligase (Biolabs). An aliquot of the ligation reaction was used to transform E. coli DH5α competent cells. In order to ascertain the allelic phase for each patient, the plasmid DNA from at least five independent clones was purified and studied by sequence analysis by using the same primers as described above.
Abbreviations
| CJD | = | sporadic Creutzfeldt-Jakob disease |
| gCJD | = | genetic Creutzfeldt-Jakob disease |
| GSS | = | Gerstmann-Straüssler-Sheinker syndrome |
| FFI | = | fatal familial insomnia |
| Ct | = | cycle threshold |
Figures and Tables
Figure 1 PRNP genotypes vs Ct (M129-178/200 wildtype, circles; M129-178/200 mutant, squares; V129-178/200 wildtype, triangles and V129-178/200 mutant, rhombus and negative controls, NC).
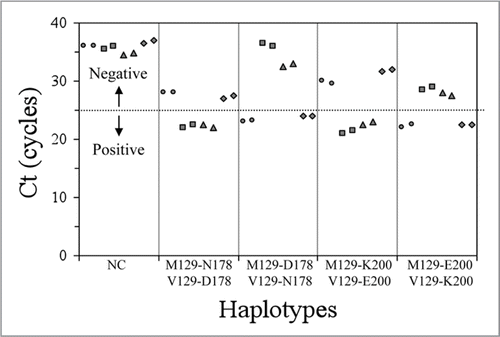
Figure 2 Amplification plots (cycle vs ΔRn) in a StepOne Real-Time PCR System of the different PRNP genotypes (M129-178/200 wildtype in red, M129-178/200 mutant in green, V129-178/200 wildtype in blue and V129-178/200 mutant in magenta). ΔRn is an indicator of the magnitude of the signal generated by the PCR reaction.
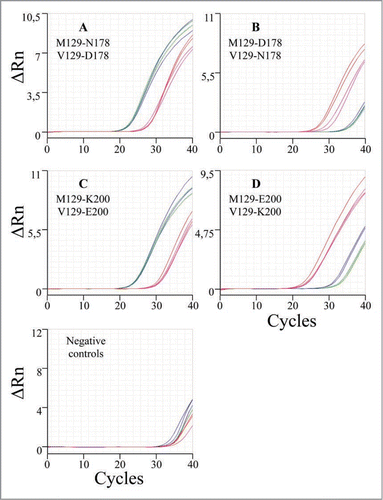
Figure 3 Performance of the allelic discrimination protocol for the M129V-D178N genetic background as a function of the initial DNA amount vs Ct (M129-178 wildtype, circles; M129-178 mutant, squares; V129-178 wildtype, triangles; V129-178 mutant, rhombus).
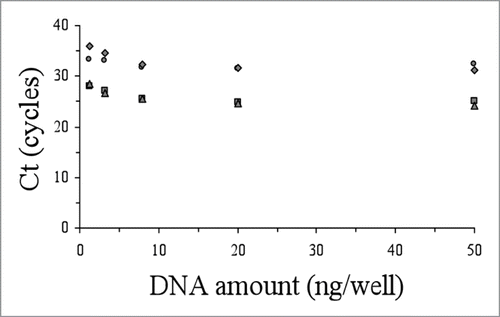
Figure 4 Performance of the allelic discrimination protocol for the M129V-E200K genetic background as a function of the initial DNA amount vs Ct (M129-200 wildtype, circles; M129-200 mutant, squares; V129-200 wildtype, triangles; V129-200 mutant, rhombus).
Table 1 Nucleotide sequence and characteristics of the primers used for PRNP genotyping
Table 2 Reaction mixtures and lengths of the amplification products expected for PRNP genotyping
References
- Prusiner SB. Molecular biology of prion diseases. Science 1991; 252:1515 - 1522
- Basler K, Oesch B, Scott M, Westaway D, Walchli M, Groth DF, et al. Scrapie and cellular PrP isoforms are encoded by the same chromosomal gene. Cell 1986; 46:417 - 428
- Aguzzi A, Baumann F, Bremer J. The prion's elusive reason for being. Annu Rev Neurosci 2008; 31:439 - 477
- Mead S. Prion disease genetics. Europ J Hum Genet 2006; 14:273 - 281
- Kovacs GG, Puopolo M, Ladogana A, Pocchiari M, Budka H, van Duijn C, et al. Genetic prion disease: the EUROCJD experience. Hum Genet 2005; 118:166 - 174
- Goldfarb LG, Brown P, Haltia M, Cathala F, McCombie WR, Kovanen J, et al. Creutzfeldt-Jakob disease cosegregates with the codon 178-asn PRNP mutation in Families of European origin. Ann Neurol 1992; 31:274 - 281
- Zarranz JJ, Digon A, Atarés B, Rodríguez-Martínez AB, Arce A, Carrera N, et al. Phenotypic variability in familial prion diseases due to the D178N mutation. J Neurol Neurosurg Psychiatry 2005; 76:1491 - 1496
- Goldfarb LG, Mitrová E, Brown P, Toh BK, Gajdusek DC. Mutation in codon 200 of scrapie amyloid protein gene in two clusters of Creutzfeldt-Jakob disease in Slovakia. Lancet 1990; 336:514 - 515
- Puoti G, Rossi G, Giaccone G, Awan T, Lievens PM, Defanti CA, et al. Polymorphism at codon 129 of PRNP affects the phenotypic expression of Creutzfeldt-Jakob disease linked to E200K mutation. Ann Neurol 2000; 48:269 - 270
- Hainfellner JA, Parchi P, Kitamoto T, Jarius C, Gambetti P, Budka H. A novel phenotype in familial Creutzfeldt-Jakob disease: prion protein gene E200K mutation coupled with valine at codon 129 and type 2 protease-resistant prion protein. Ann Neurol 1999; 45:812 - 816
- Collinge J, Palmer MS. Prion diseases. Curr Opin Genet Dev 1992; 2:448 - 454
- Collinge J. Molecular neurology of prion disease. J Neurol Neurosurg Psychiatry 2005; 76:906 - 919
- Sambrook J, Russell DW. Molecular cloning 2001; New York CSHL Press, Cold Spring Harbor
- Young K, Clark HB, Piccardo P, Dlouhy SR, Ghetti B. Gerstmann-Sträussler-Scheinker disease with the PRNP P102L mutation and valine at codon 129. Brain Res Mol Brain Res 1997; 44:147 - 150
- Asante EA, Gowland I, Grimshaw A, Linehan JM, Smidak M, et al. Absence of spontaneous disease and comparative prion susceptibility of transgenic mice expressing mutant human prion proteins. J Gen Virol 2009; 90:546 - 558