Abstract
The best known attribute of the prion protein (PrP) is its tendency to misfold into a rogue isoform. Much less understood is how this misfolded isoform causes deadly brain illnesses. Although neurodegeneration in prion disease is often seen as the result of abnormal PrP function, amazingly little is known about the normal, physiological role of PrP. In particular, the absence of obvious phenotypes in PrP knockout mice has prevented scientists from answering this important question. Using knockdown approaches, we previously produced clear PrP loss-of-function phenotypes in zebrafish. Analysis of these phenotypes revealed that PrP can modulate E-cadherin-based cell adhesion, thereby controlling essential morphogenetic cell movements in the early embryo. Our data also showed that PrP itself can elicit homophilic cell-cell adhesion and trigger intracellular signaling via Src-related kinases. Here we discuss the use of the zebrafish in prion biology, and how these findings may advance our understanding of the roles of PrP in health and disease.
Introduction
In 1982, Stanley Prusiner reported that novel proteinaceous infectious particles—prions—could replicate without nucleic acids and transmit deadly neurological diseases.Citation1 Three years later, the unconventional pathogens were found to be composed of a host-derived substance, the prion protein (PrP).Citation2,Citation3 Interestingly, this cell-surface molecule is produced in many tissues as a normal constituent of the cell. What sets it apart from other proteins, though, is its remarkable ability to misfold into a self-propagating conformation with the tendency to aggregate and form infectious prions.Citation4 Curiously, although prions may accumulate in different cell types,Citation5 it is only in neurons where they cause extensive cell death, the pathological landmark of neurodegenerative disorders like Creutzfeldt-Jakob disease (CJD) in humans and bovine spongiform encephalopathy (BSE) in cattle.Citation6
Perhaps the most elusive questions in prion biology concern the physiological role of PrP and the cellular mechanisms by which prions cause brain damage. While these two matters may seem unrelated at first glance, experimental evidence suggests that they are causally related. For instance, transgenic mice in which PrP was modified to prevent its attachment to the plasma membrane have been shown to replicate prions without developing prion disease.Citation7 This and other compelling studies have revealed that an activity of PrP at the cell surface is necessary for prion-induced neurodegeneration to occur. Hence, the physiological function of PrP may hold the key to the mystery of prion pathogenesis.
Unfortunately, ascertaining the natural role of PrP has proven to be an arduous and deceiving task. Based for the most part on in vitro studies, a plethora of dissimilar functions have been proposed for PrP, including cytoprotection from apoptosis and oxidative stress, copper metabolism, neurogenesis, lymphocyte activation, axonal growth, synapse formation and maintenance, hematopoietic stem cell self-renewal, signal transduction and cell adhesion.Citation8 Nevertheless, the physiological relevance of most of these putative roles is not clear, nor is their mechanistic relationship to neurodegeneration. Analysis of PrP function in vivo has been even less rewarding. Beyond a few subtle abnormalities, PrP knockout mice develop and behave rather normally, their only clear “phenotype” being their resistance to prion infection.Citation9 On the other hand, the use of simpler genetic models like nematodes and flies has contributed only limited information to the subject, arguably because these animals lack PrP. Interestingly, non-homologous prion proteins have been extensively studied in yeast and fungi. However, the similarity of these proteins to PrP is confined to their ability to misfold and replicate and thus, they are not suited for PrP functional analysis.
Breaking the “No Phenotype” Spell
We chose to investigate the roles of PrP in the zebrafish because of the many experimental advantages it has over mammalian and invertebrate organisms. Zebrafish embryos develop externally and are optically clear, making it feasible to carry out detailed cellular analyses and genetic manipulations in a species that is evolutionarily closer to mammals than flies or worms. Moreover, our work has shown that zebrafish have bona fide PrPs expressed at high levels in the adult and developing brain.Citation10 These duplicated proteins, PrP-1 and PrP-2, share key biochemical properties with their mammalian counterparts, such as protein domain composition, patterns of N-glycosylation, and attachment to the plasma membrane via a GPI-anchor.Citation10,Citation11
In a recent study, we used the zebrafish model to show that PrP provides cellular signals that regulate cell communication in vivo.Citation12 In these experiments, embryonic expression of PrP-1 or -2 was knocked down by microinjecting morpholino antisense oligonucleotides into freshly fertilized eggs. The resulting embryos (morphants) exhibited dramatic morphological defects. Knockdown of PrP-1 prevented embryos from carrying out gastrulation and led to early developmental arrest. In contrast, PrP-2 depletion did not affect gastrulation but produced embryos with severely malformed heads and eyes. Remarkably, the PrP-1 arrested phenotype could be rescued not only by PrP-1, but also partially by PrP-2 and even mouse PrP mRNAs, strongly supporting the notion of functional homology between fish and mammalian PrPs. In addition, the rescue experiments indicate that although PrP-1 and -2 are deployed in different developmental contexts, they share a basic biological activity with mouse PrP. To the best of our knowledge, these results are the first experimental demonstration that the absence of PrP can cause dramatic physiological abnormalities in a living animal. At the same time, the zebrafish data pose an intriguing paradox: if fish and mammalian PrPs share a conserved function, why is the phenotype of the knockout mouse so subtle?
A simple answer would be that PrP is not essential for mammalian embryogenesis. This might be, however, too facile an explanation, considering the fact that the role of PrP in the mouse gastrula has been overlooked by prion researchers. Alternatively, as proposed by us and others,Citation9 if PrP indeed plays an important role in the mouse embryo, the knockout phenotype could become masked by genetic compensation or developmental plasticity. But, why would such compensatory mechanisms be activated in mice and not in zebrafish? The reason may be technical. Knockout mice are derived from cultured embryonic stem cells, which are artificially selected for removal of the targeted locus by homologous recombination. Deleterious PrP knockout effects may not appear in the embryo because only cultured cells that are able to activate compensatory mechanisms would survive and be used to generate the knockout mouse. In knockdown fish embryos, however, translation is sterically hindered but the physical locus remains intact and transcriptionally active. Thus, loss-of-function can be directly observed and no individual cells are selected because the embryo behaves as a single entity. This hypothetical argument implies that clear PrP phenotypes might become visible in mice only upon replacement of the PrP gene with truncated copies. In fact, several of such experiments have already been reported and support this view.Citation6,Citation8 Also, it may be of interest to identify compensatory mechanisms in mice by analyzing gene expression profiles in PrP knockout embryonic stem cells and embryos.
The Road from Phenotype to Cellular Function
PrP-1 and -2 serve very different purposes during zebrafish embryogenesis. While early ubiquitous expression of PrP-1 is essential for gastrulation, restricted expression of PrP-2 in the developing nervous system is required for the proper formation of neural structures. Yet, our rescue experiments indicate that the two proteins are functionally related. So, which single cellular function could account for such diverse developmental roles?
To address this question, we first analyzed the patterns of PrP subcellular localization in cultured mammalian cells and zebrafish embryos. We noticed that fluorescently tagged versions of zebrafish and mouse PrPs accumulated locally at cell-cell contacts, and that the accumulation was dependent on the expression of PrP on the surface of both cells forming the contact. This crucial observation led us to hypothesize that PrPs on apposing cell membranes may interact in trans, thereby influencing the stability of cell-cell contacts. If so, the PrP-1 and -2 knockdown phenotypes would be explained by defects in cell-cell communication. To verify this, we focused our analysis on the cellular and molecular characterization of the PrP-1 phenotype, given the relative simplicity and ease of manipulation of the early embryo. Detailed morphological examination showed that the gastrulation arrest was preceded by a marked decrease in tissue integrity, due to the progressive loss of cell-cell adhesion. Conversely, in rescue experiments, cell-cell adhesion could be restored by adding exogenous PrP, which localized preferentially at cell-cell contact sites. In addition, when PrP-1 morphant cells were transplanted into control embryos, they failed to establish cell contacts, indicating that the adhesion defect was cell autonomous and could not be reverted by the presence of PrP-1 in the host embryo. These experiments confirmed that the accumulation of PrP-1 at cell-cell contacts is required for the maintenance of embryonic cell adhesion. But then, does this equal to saying that PrP-1 is an adhesion molecule?
Not entirely. In the early embryo, cell-cell adhesion relies largely on the maintenance of adherens junctions. These specialized structures are supported by Ca+2-dependent, homophilic interactions between E-cadherin molecules on neighbouring cell membranes.Citation13 Therefore, we reasoned that PrP-1 could influence embryonic cell adhesion by modulating the function of E-cadherin. To test this notion, control and PrP-1 morphant embryos were dissociated to single-cell suspensions and the cells were allowed to reaggregate with or without Ca+2. The adhesive properties of the cells were measured by their ability to form large (E-cadherin-dependent) or small (E-cadherin-independent) cell clusters. Notably, in the presence of Ca+2, PrP-1 knockdown abolished the formation of large cell clusters and visibly reduced the number of small cell clusters. In the absence of Ca+2, large cell clusters rarely formed but PrP-1 knockdown still caused a significant decrease in the number of small cell clusters. Similarly, knocking down PrP-1 and E-cadherin simultaneously had a considerably greater effect on cell adhesion than each of the single knockdowns alone, indicating a synergistic genetic interaction between the two molecules. We concluded that PrP-1 contributes to embryonic cell-cell adhesion not only through its own adhesive properties but also indirectly, via the regulation of E-cadherin. The importance of these roles of PrP goes beyond the maintenance of embryonic tissue integrity. For instance, during blastula and gastrula stages, the fine modulation of E-cadherin-mediated cell adhesion is crucial to control the complex morphogenetic cell movements that give rise to the germ layers. Accordingly, we have demonstrated that PrP-1 morphant embryos undergo gastrulation arrest because they fail to carry out a specific morphogenetic cell movement known as radial intercalation.
How exactly does PrP-1 modulate E-cadherin activity? The control of E-cadherin function is a rather complex phenomenon involving diverse mechanisms, such as the regulation of gene transcription, contact-induced conformational changes, posttranslational cleavage, phosphorylation of catenins, internalization by endocytosis, as well as lysosomal and proteasomal degradation.Citation13 We have shown that PrP-1 knockdown disrupts the normal membrane localization of E-cadherin and its associated molecule β-catenin, as well as the organization of the actin cytoskeleton. Furthermore, our analysis of PrP-1 morphant embryos revealed an abnormal accumulation of E-cadherin in intracellular vesicles, along with a sharp reduction in the levels of mature, membrane-bound E-cadherin. These results indicate that PrP-1 influences the processing of E-cadherin, and its transport to or from the plasma membrane. Are then PrP and E-cadherin physical interaction partners? This does not seem to be the case, as they have been shown to co-localize but not to physically interact in cell junctions of human enterocytes.Citation14 In our experiments, the limited co-localization of PrP-1 and E-cadherin in zebrafish blastomeres also argues against an obligatory physical interaction. Instead, we think that the modulation of E-cadherin by PrP-1 is likely to occur indirectly, via signaling. In fact, when we allowed dissociated blastomeres to reaggregate, we observed that the local accumulation of E-cadherin at PrP-1-mediated cell-cell contacts was accompanied by the local activation of the Src-related tyrosine kinase Fyn. Altogether, these data suggest that PrP-1 sets off an intracellular signaling cascade, which ultimately may control the trafficking, endocytosis and degradation of cadherin/catenin complexes, as well as the stability of the actin cytoskeleton.
Because of the genetic and functional complexities of the living embryo, we also have used a simplified cell culture assay to confirm that PrPs possess their own, intrinsic adhesive and signaling properties. Drosophila Schneider 2 (S2) cells lack endogenous PrP, do not express adhesion molecules, and therefore grow as single-cell suspensions. However, when we transfected them with mouse, zebrafish, frog or chicken PrP constructs, they acquired the ability to build cell clusters and accumulate PrP at cell-cell contacts. These effects were accompanied by the local accumulation of activated Src-kinases and tyrosine-phosphorylated proteins at cell-cell contact sites. Intriguingly, cell aggregation and intracellular signaling were also elicited among cells separately transfected with mouse and fish PrPs, revealing that PrP trans-interactions are very conserved and can take place even across a wide species range. If, as thought, PrP-mediated signals play a key role in prion pathogenesis, the observed interaction between fish and mammalian PrPs raises the need to assess whether exposure of fish to mammalian prions would lead to the generation of infectious fish prions.
Implications and Future Directions
What conclusions can be drawn from these experiments? First and foremost, our study shows that the lack of PrP-1 in zebrafish produces a clear in vivo phenotype amenable to molecular characterization. In particular, the finding that this phenotype can be partially reverted by the mouse protein underscores the functional similarities between fish and mammalian PrPs. In addition, our data provide a mechanistic explanation for the phenotype at the cellular level, namely, the impairment of morphogenetic cell movements due to the loss of cell-cell adhesion. Our results also indicate that the molecular basis for this defect is the role of PrP as a modulator of Ca+2-dependent cell adhesion, through the regulation of E-cadherin activity. Finally, we demonstrated that PrP itself can also mediate Ca+2-independent homophilic cell adhesion and trigger phosphorylation signals, even across distantly related species.
Could the zebrafish findings be of potential interest to mainstream prionologists? Can the zebrafish be used to model prion disease? Some may rightfully argue that fish PrPs need yet to earn their name, in light of the fact that infectious (PrP) prions so far have only been reported in mammals. Nevertheless, it also must be acknowledged that the possibility of prion diseases in fish has been examined only superficially and that studies on this subject are still very far from reaching the level of sophistication achieved in the mouse prion field. The generation of zebrafish PrP transgenic lines might help change this, but until then, it may be premature to rule out the possibility of fish—particularly farmed fish—acquiring and transmitting prion diseases. After all, the scenario of a mad cow disease epidemic probably would have seemed audacious 30 years ago!
Regardless of whether piscine prions would pose a risk to public health, the study of fish PrPs may prove very insightful. For example, research in zebrafish embryos could help identify PrP functions related to the onset of mammalian prion disease. This, in turn, would facilitate the search for novel therapeutic targets to block neurodegeneration. Furthermore, the zebrafish could be turned into a simple and cost-efficient tool for drug screening. But how would one go from malformed fish embryos to adult mad cows? On one hand, the existence of two PrPs in the zebrafish provides a unique opportunity to separately address the molecular basis of PrP function (PrP-1 in the early gastrula), and its physiological relevance in the brain (PrP-2 in developing neurons). On the other hand, it has been pointed out elsewhere that our findings are consistent with previous observations pertaining proposed roles of mammalian PrP in cell-cell interactions, Src-based signaling and neurite outgrowth, as well as in neural development.Citation15 Indeed, there are some striking similarities between the molecular networks active in the zebrafish gastrula and in the mammalian brain. For example, besides their known involvement in embryonic cell adhesion, cadherins and catenins play key roles in mammalian synaptogenesis and synaptic plasticity.Citation16 Likewise, alterations in the stability of β-catenin have been reported to increase neuronal apoptosis during Alzheimer's disease (AD).Citation17 More recently, PrP was found to function as a receptor for amyloid-beta oligomers, and to mediate the AD-associated impairment of synaptic plasticity.Citation18 Thus, elucidating the mechanisms of PrP-mediated signaling in the zebrafish might help clarify the common molecular basis of these neurodegenerative disorders.
What could be the cellular signals induced by PrP homophilic interactions at the cell surface? How would they exert control over E-cadherin and the actin cytoskeleton? A wealth of experimental evidence suggests that Src-related tyrosine kinases may be central to these matters. For example, it is known that Src-related kinases modulate the stability of adherens junctions by directly phosphorylating cadherin/catenin complexes.Citation13,Citation19,Citation20 Moreover, p120 catenin (also a target of Src-related kinases) can directly bind E-cadherin and effectively control its function, as well as the activity of small GTPases.Citation21 Some of these, like RhoA and its relatives Rac and Cdc42, are important regulators of actin dynamics and cell adhesion,Citation22 whereas others, like Rab 5 and Rab11, mediate the endocytosis and exocytosis of E-cadherin, respectively.Citation23
Most relevant to this discussion is the finding that Src-related kinases like Fyn and Yes can signal via RhoA to control gastrulation cell movements in the zebrafish.Citation24 The function of Rho is mediated by its downstream effector molecules Rok2 and Diaphanous, which directly affect cell morphology and migration in the gastrula.Citation25–Citation28 This signaling cascade is regulated by C-terminal Src kinase (CSK) and various protein tyrosine phosphatases (PTPs), which act directly upstream of Fyn and Yes.Citation29–Citation31 In addition, PTPs can also control cadherin-based cell adhesionCitation20 and cell movement,Citation32 as well as axon guidance and neurite outgrowth.Citation33,Citation34 Finally, knockdown of these molecules in the zebrafish produces gastrulation defects partly related to those we observe in PrP-1 knockdown embryos.
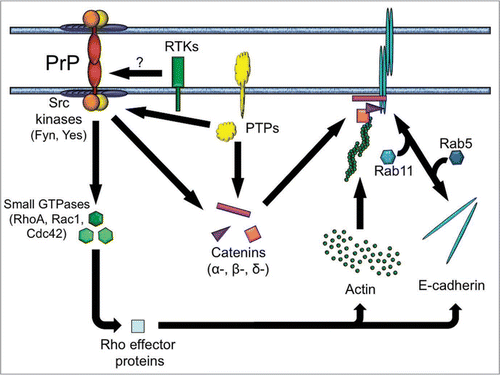
Based on these data, we constructed a hypothetical scenario describing some of the molecular pathways potentially downstream of PrP (). In particular, the model provides testable hypotheses concerning the role of tyrosine kinases, catenins and small GTPases in PrP-mediated cell-cell communication. It remains to be clarified how these molecules and other associated pathways may contribute to prion disease. Putting together the pieces of this complex puzzle will certainly be an exciting challenge. The zebrafish may be for now the rookie of prion biology but it has already shown that PrPs are, after all, proteins with a purpose.
Figures and Tables
Figure 1 A proposed role of PrP in cell-cell communication. Homophilic trans-interactions between PrP molecules elicit contact formation and signal transduction by Src-related tyrosine kinases, leading to the correct assembly and positioning of E-cadherin adhesion complexes, as well as to remodeling of the actin cytoskeleton via small GTPases. These processes may be further modulated by additional molecules, including catenins (α-, β- and δ-catenin), protein tyrosine phosphatases (PTPs) and external cues via receptor tyrosine kinases (RTKs). Although the model assumes that PrP itself is capable of eliciting a signal across the plasma membrane, it does not exclude the possibility that PrP may also signal through a cis-interacting partner. Arrows do not imply unidirectionality.
Note
In a recent study, Salta et al. have provided the first evidence of neurodegeneration and plaque-like aggregates in the brains of fish fed with bovine and ovine prions.Citation35 These data highlight the need to ascertain the occurrence, transmissibility and infectivity of fish prions.
References
- Prusiner SB. Novel proteinaceous infectious particles cause scrapie. Science 1982; 216:136 - 144
- Oesch B, Westaway D, Walchli M, McKinley MP, Kent SB, Aebersold R, et al. A cellular gene encodes scrapie PrP 27–30 protein. Cell 1985; 40:735 - 746
- Chesebro B, Race R, Wehrly K, Nishio J, Bloom M, Lechner D, et al. Identification of scrapie prion protein-specific mRNA in scrapie-infected and uninfected brain. Nature 1985; 315:331 - 333
- Weissmann C. The state of the prion. Nat Rev Microbiol 2004; 2:861 - 871
- Bosque PJ, Ryou C, Telling G, Peretz D, Legname G, DeArmond SJ, et al. Prions in skeletal muscle. Proc Natl Acad Sci USA 2002; 99:3812 - 3817
- Aguzzi A, Baumann F, Bremer J. The prion's elusive reason for being. Annu Rev Neurosci 2008; 31:439 - 477
- Chesebro B, Trifilo M, Race R, Meade-White K, Teng C, LaCasse R, et al. Anchorless prion protein results in infectious amyloid disease without clinical scrapie. Science 2005; 308:1435 - 1439
- Westergard L, Christensen HM, Harris DA. The cellular prion protein (PrP(C)): its physiological function and role in disease. Biochim Biophys Acta 2007; 1772:629 - 644
- Steele AD, Lindquist S, Aguzzi A. The prion protein knockout mouse: a phenotype under challenge. Prion 2007; 1:83 - 93
- Rivera-Milla E, Oidtmann B, Panagiotidis CH, Baier M, Sklaviadis T, Hoffmann R, et al. Disparate evolution of prion protein domains and the distinct origin of Doppel- and prion-related loci revealed by fish-to-mammal comparisons. Faseb J 2006; 20:317 - 319
- Rivera-Milla E, Stuermer CA, Malaga-Trillo E. An evolutionary basis for scrapie disease: identification of a fish prion mRNA. Trends Genet 2003; 19:72 - 75
- Málaga-Trillo E, Solis GP, Schrock Y, Geiss C, Luncz L, Thomanetz V, et al. Regulation of embryonic cell adhesion by the prion protein. PLoS Biol 2009; 7:1000055
- Nelson WJ. Regulation of cell-cell adhesion by the cadherin-catenin complex. Biochem Soc Trans 2008; 36:149 - 155
- Morel E, Fouquet S, Chateau D, Yvernault L, Frobert Y, Pincon-Raymond M, et al. The cellular prion protein PrPc is expressed in human enterocytes in cell-cell junctional domains. J Biol Chem 2004; 279:1499 - 1505
- Chiesa R, Harris DA. Fishing for prion protein function. PLoS Biol 2009; 7:75
- Arikkath J, Reichardt LF. Cadherins and catenins at synapses: roles in synaptogenesis and synaptic plasticity. Trends Neurosci 2008; 31:487 - 494
- Zhang Z, Hartmann H, Do VM, Abramowski D, Sturchler-Pierrat C, Staufenbiel M, et al. Destabilization of beta-catenin by mutations in presenilin-1 potentiates neuronal apoptosis. Nature 1998; 395:698 - 702
- Lauren J, Gimbel DA, Nygaard HB, Gilbert JW, Strittmatter SM. Cellular prion protein mediates impairment of synaptic plasticity by amyloid-beta oligomers. Nature 2009; 457:1128 - 1132
- Lilien J, Balsamo J. The regulation of cadherin-mediated adhesion by tyrosine phosphorylation/dephosphorylation of beta-catenin. Curr Opin Cell Biol 2005; 17:459 - 465
- Sallee JL, Wittchen ES, Burridge K. Regulation of cell adhesion by protein-tyrosine phosphatases: II. Cell-cell adhesion. J Biol Chem 2006; 281:16189 - 16192
- Alema S, Salvatore AM. p120 catenin and phosphorylation: Mechanisms and traits of an unresolved issue. Biochim Biophys Acta 2007; 1773:47 - 58
- Reynolds AB, Roczniak-Ferguson A. Emerging roles for p120-catenin in cell adhesion and cancer. Oncogene 2004; 23:7947 - 7956
- Lock JG, Stow JL. Rab11 in recycling endosomes regulates the sorting and basolateral transport of E-cadherin. Mol Biol Cell 2005; 16:1744 - 1755
- Jopling C, den Hertog J. Fyn/Yes and non-canonical Wnt signalling converge on RhoA in vertebrate gastrulation cell movements. EMBO Rep 2005; 6:426 - 431
- Lai SL, Chan TH, Lin MJ, Huang WP, Lou SW, Lee SJ. Diaphanous-related formin 2 and profilin I are required for gastrulation cell movements. PLoS ONE 2008; 3:3439
- Lai SL, Chang CN, Wang PJ, Lee SJ. Rho mediates cytokinesis and epiboly via ROCK in zebrafish. Mol Reprod Dev 2005; 71:186 - 196
- Marlow F, Topczewski J, Sepich D, Solnica-Krezel L. Zebrafish Rho kinase 2 acts downstream of Wnt11 to mediate cell polarity and effective convergence and extension movements. Curr Biol 2002; 12:876 - 884
- Zhu S, Liu L, Korzh V, Gong Z, Low BC. RhoA acts downstream of Wnt5 and Wnt11 to regulate convergence and extension movements by involving effectors Rho Kinase and Diaphanous: Use of zebrafish as an in vivo model for GTPase signaling. Cell Signal 2006; 18:359 - 372
- Jopling C, Hertog J. Essential role for Csk upstream of Fyn and Yes in zebrafish gastrulation. Mech Dev 2007; 124:129 - 136
- Roskoski R Jr. Src kinase regulation by phosphorylation and dephosphorylation. Biochem Biophys Res Commun 2005; 331:1 - 14
- Jopling C, van Geemen D, den Hertog J. Shp2 knockdown and Noonan/LEOPARD mutant Shp2-induced gastrulation defects. PLoS Genet 2007; 3:225
- Schoenwaelder SM, Petch LA, Williamson D, Shen R, Feng GS, Burridge K. The protein tyrosine phosphatase Shp-2 regulates RhoA activity. Curr Biol 2000; 10:1523 - 1526
- Holland SJ, Peles E, Pawson T, Schlessinger J. Cell-contact-dependent signalling in axon growth and guidance: Eph receptor tyrosine kinases and receptor protein tyrosine phosphatase beta. Curr Opin Neurobiol 1998; 8:117 - 127
- Yang T, Yin W, Derevyanny VD, Moore LA, Longo FM. Identification of an ectodomain within the LAR protein tyrosine phosphatase receptor that binds homophilically and activates signalling pathways promoting neurite outgrowth. Eur J Neurosci 2005; 22:2159 - 2170
- Salta E, Panagiotidis C, Teliousis K, Petrakis S, Eleftheriadis E, Arapoglou F, et al. Evaluation of the possible transmission of BSE and scrapie to gilthead sea bream (Sparus aurata). PLoS ONE 2009; 4:6175