Abstract
In transmissible spongiform encephalopathies (TSE or prion diseases) such as sheep scrapie, bovine spongiform encephalopathy and human Creutzfeldt-Jakob disease, normally soluble and protease-sensitive prion protein (PrP-sen or PrPC) is converted to an abnormal, insoluble and protease-resistant form termed PrP-res or PrPSc. PrP-res/PrPSc is believed to be the main component of the prion, the infectious agent of the TSE/prion diseases. Its precursor, PrP-sen, is anchored to the cell surface at the C-terminus by a co-translationally added glycophosphatidyl-inositol (GPI) membrane anchor which can be cleaved by the enzyme phosphatidyl-inositol specific phospholipase (PIPLC). The GPI anchor is also present in PrP-res, but is inaccessible to PIPLC digestion suggesting that conformational changes in PrP associated with PrP-res formation have blocked the PIPLC cleavage site. Although the GPI anchor is present in both PrP-sen and PrP-res, its precise role in TSE diseases remains unclear primarily because there are data to suggest that it both is and is not necessary for PrP-res formation and prion infection.
In transmissible spongiform encephalopathies (TSE or prion diseases) such as sheep scrapie, bovine spongiform encephalopathy and human Creutzfeldt-Jakob disease, normally soluble and protease-sensitive prion protein (PrP-sen or PrPC) is converted to an abnormal, insoluble and protease-resistant form termed PrP-res or PrPSc. PrP-res/PrPSc is believed to be the main component of the prion, the infectious agent of the TSE/prion diseases. Its precursor, PrP-sen, is anchored to the cell surface at the C-terminus by a co-translationally added glycophosphatidyl-inositol (GPI) membrane anchor which can be cleaved by the enzyme phosphatidyl-inositol specific phospholipase (PIPLC). The GPI anchor is also present in PrP-res, but is inaccessible to PIPLC digestion suggesting that conformational changes in PrP associated with PrP-res formation have blocked the PIPLC cleavage site.Citation1 Although the GPI anchor is present in both PrP-sen and PrP-res, its precise role in TSE diseases remains unclear primarily because there are data to suggest that it both is and is not necessary for PrP-res formation and prion infection.
In tissue culture cells infected with mouse scrapie, PrP-res formation occurs at the cell surface and/or along the endocytic pathwayCitation2–Citation4 and may be dependent upon the membrane environment of PrP-sen. For example, localization via the GPI anchor to caveolae-like domains favors PrP-res formationCitation5 while substitution of the GPI anchor addition site with carboxy termini favoring transmembrane anchored PrP-sen inhibits formation of PrP-res.Citation5,Citation6 Other studies have shown that localization of both PrP-sen and PrP-res to lipid rafts, cholesterol and sphingolipid rich membrane microdomains where GPI anchored proteins can be located, is important in PrP-res formation.Citation6–Citation9
However, there are also data which suggest that such localization is not necessarily essential for PrP-res formation. Anchorless PrP-sen isolated from cells by immunoprecipitation or wild-type PrP-sen purified by immunoaffinity column followed by cation exchange chromatography are efficiently converted into PrP-res in cell-free systems.Citation10,Citation11 Furthermore, recombinant PrP-sen derived from E. coli, which has no membrane anchor or glycosylation, can be induced to form protease-resistant PrP in vitro when reacted with prion-infected brain homogenates.Citation12–Citation14 Finally, in at least one instance, protease-resistant recombinant PrP-res generated in the absence of infected brain homogenate was reported to cause disease when inoculated into transgenic mice.Citation15
The data concerning the role of the PrP-sen GPI anchor in susceptibility to TSE infection are similarly contradictory. Transgenic mice expressing anchorless mouse PrP-sen are susceptible to infection with mouse scrapie and accumulate both PrP-res and prion infectivity.Citation16 Thus, the GPI anchor is clearly not needed for PrP-res formation or productive TSE infection in vivo. However, we recently published data demonstrating that, in vitro, anchored PrP-sen is in fact required to persistently infect cells.Citation17 Given that anchorless PrP-sen is not present on the cell surface but is released into the cell medium, we speculated that the differences between the in vitro and in vivo data were related to the location of PrP-res formation. In the mice expressing anchorless PrP-sen, environments conducive to PrP-res formation are present in certain areas of the complex extracellular milieu of the brain where anchorless, secreted PrP-sen can accumulate and come into contact with PrP-res from the infectious inoculum. Since similar environments are missing in vitro, any PrP-res formation in cells expressing anchorless PrP-sen must be cell-associated. While this explanation addresses how extracellular PrP-res could be generated in an unusual transgenic mouse model of TSE infection, it does not really help to define how the GPI anchor is involved in normal prion infection of a cell.
As with other infectious organisms such as viruses, TSE infection can be roughly divided into three steps: uptake, replication and spread. Over the last several years, data derived from new techniques as well as new cell lines susceptible to prion infection have increased our knowledge of some of the basic events that occur during each of these steps. In order to try to tease out the role of the GPI anchor in normal TSE pathogenesis, it is therefore useful to consider the process of TSE infection of a cell and how the GPI anchor might be involved in each stage.
In a conventional viral infection, binding and uptake of the virus is essential to establish infection. Studying PrP-res uptake has been complicated by the lack of an antibody that can specifically distinguish PrP-res from PrP-sen in live cells and by the difficulty of detecting the input PrP-res from the PrP-res made de novo by the cell. Recently, however, several groups have been able to study PrP-res uptake using input PrP-res that was either fluorescently labeledCitation18–Citation20 or tagged with the epitope to the monoclonal antibody 3F4,Citation21 or cell lines that express little or no PrP-sen.Citation19,Citation21–Citation23 The data show that PrP-res uptake is independent of scrapie strain or cell type but is influenced by the PrP-res microenvironment as well as PrP-res aggregate size.Citation21 Importantly, these studies demonstrated that PrP-sen expression was not required.Citation19,Citation21–Citation23 Given these data, it is clear that GPI anchored PrP-sen is not involved in the initial uptake of PrP-res into the cell.
The next stage of prion infection involves replication of infectivity which is typically assayed by following cellular PrP-res formation. Once again, however, the issue of how to distinguish PrP-res in the inoculum from newly formed PrP-res in the cells has made it difficult to study the early stages of prion replication. To overcome this difficulty, we developed a murine tissue culture system that utilizes cells expressing mouse PrP-sen tagged with the epitope to the 3F4 antibody (Mo3F4 PrP-sen).Citation24 Wild-type mouse PrP does not have this epitope. As a result, following exposure to an infected mouse brain homogenate, de novo PrP-res formation can be followed by assaying for 3F4 positive PrP-res. Our studies showed that there were two stages of PrP-res formation: (1) an initial acute burst within the first 96 hours post-infection that was cell-type and scrapie strain independent and, (2) persistent PrP-res formation (i.e., formation of PrP-res over multiple cell passages) that was dependent on cell-type and scrapie strain and associated with long-term infection.Citation24 Acute PrP-res formation did not necessarily lead to persistent PrP-res formation suggesting that other cell-specific factors or processes are needed for PrP-res formation to persist.Citation24
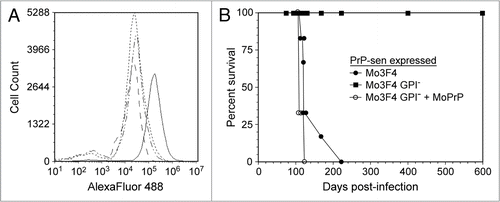
When cells expressing Mo3F4 PrP-sen without the GPI anchor (Mo3F4 GPI-PrP-sen) were exposed to mouse scrapie infected brain homogenates, GPI negative, 3F4 positive PrP-res (Mo3F4 GPI-PrP-res) was detected within 96 hours indicating that acute PrP-res formation had occurred.Citation17 Thus, despite the fact that Mo3F4 GPI-PrP-sen is not expressed on the cell surfaceCitation16 (), it was still available for conversion to PrP-res. These results are consistent with data from cell-free systems and demonstrate that, at least acutely, membrane anchored PrP is not necessary for PrP-res formation in a cell.
In terms of persistent PrP-res formation, however, our data suggest that the GPI anchor is important. Despite an initial burst of PrP-res formation within the first 96 hours post-infection, Mo3F4 GPI-PrP-res was not observed following passage of the cells nor did the cells become infected. This effect was not due either to resistance of the cells to scrapie infection or to an inability of the scrapie strain used to infect cells. When the same cells expressed anchored Mo3F4 PrP-sen and were exposed to the same mouse scrapie strain, both acute and persistent PrP-res formation were detected and the cells were persistently infected with scrapie ().Citation17 Taken together, these data demonstrate that cells expressing anchorless PrP-sen do not support persistent PrP-res formation. Furthermore, the data strongly suggest that GPI-anchored PrP-sen is required during the transition from acute to persistent scrapie infection. In support of this hypothesis, the resistance of cells expressing Mo3F4 GPI-PrP-sen to persistent prion infection could be overcome if wild-type GPI anchored PrP-sen was co-expressed in the same cell. When both forms of PrP-sen were expressed, anchored and anchorless forms of PrP-res were made and the cells became persistently infected ().Citation17 Thus, the data suggest that GPI anchored PrP is necessary to establish prion infection within a cell.
How could GPI membrane anchored PrP be involved in the establishment and maintenance of persistent prion infection? Several studies have suggested that the GPI anchor is needed to localize PrP-sen to specific membrane environments where PrP-res formation is favored.Citation5–Citation8 However, if this localization was essential for PrP-res formation, GPI-PrP-sen would presumably never form PrP-res. Lacking the GPI anchor, it would not be in the correct membrane environment to support conversion. As a result, neither acute nor persistent prion infection could occur. This is obviously not the case. Transgenic mice expressing only anchorless PrP-sen generate PrP-res and can be infected with scrapie even though (1) flotation gradients showed that anchorless PrP-sen was not in the same membrane environment as anchored PrP-sen and, (2) flow cytometry analysis demonstrated that anchorless PrP-sen was not present on the cell surface.Citation16 Thus, the GPI anchor is not needed to target PrP-sen to a conversion friendly membrane environment.
Consistent with the idea that the GPI anchor is not essential for PrP-res formation, in our studies anchorless PrP-sen could form PrP-res in cells acutely infected with scrapie despite the fact that it is processed differently than anchored PrP-sen, is not present on the cell surface (), and is secreted.Citation17 Persistent formation of anchorless PrP-res only occurred when both anchored and anchorless forms of PrP were expressed in the same cell.Citation17 For this to happen both types of PrP must share a cellular compartment where PrP-res formation occurs, presumably either on the cell surface or in a specific location along the endocytic pathwayCitation2,Citation3 such as the endosomal recycling compartment.Citation4 Analysis of infected and uninfected cells co-expressing Mo3F4 GPI-PrP-sen and wild-type PrP-sen demonstrated that Mo3F4 GPI-PrP-sen was not present on the cell surface (). Thus, it is unlikely that GPI-PrP-res formation is occurring on the cell surface. We speculate that the anchored form of PrP-res encounters anchorless PrP-sen along either a secretory or endocytic pathway, allowing for the formation of anchorless PrP-res. Regardless of the precise location, the in vitro and in vivo data strongly suggest that the role of the anchor in persistent prion infection is not simply to localize PrP-sen to an environment compatible with PrP-res formation.
However, the data are consistent with the idea that GPI anchored PrP is absolutely essential for the establishment of persistent infection in vitro. This is likely related to the spread of infectivity within a culture that is necessary for maintaining a persistent infection over time. Evidence suggests that PrP-res can be transferred between cells in a variety of ways including mother-daughter cell division,Citation25 cell-to-cell contactCitation26,Citation27 and exosomes.Citation28 Tunneling nanotubes have also been hypothesized to be involved in intercellular prion spreadCitation19 and recent data suggest that spread can occur via these structures.Citation20 Any of these processes could involve the cell-to-cell transfer of PrP-res in membrane containing particles as has been observed in cell-freeCitation7 and cell-based systems.Citation29 If cell-to-cell contact were required, for example via simple physical proximity or perhaps tunneling nanotubes,Citation19,Citation20 then the conversion of cell surface PrP-sen on the naïve cell by cell surface PrP-res on the infected cell would transfer infection to the naïve cell. In this instance, GPI membrane anchored, cell surface PrP-sen would be essential as it would allow for PrP-res formation on the cell surface. If spread is via cell division, then GPI-anchored, cell surface PrP-sen would be important for its role as a precursor to PrP-res formation.Citation2 In this instance, cell surface PrP-sen would be an essential intermediate in the continuous formation of PrP-res necessary for the accumulation and amplification of PrP-res within the cell. It would also help to cycle PrP between the cell surface and intracellular compartments where PrP-res can be formed.Citation4 In either case, GPI-anchored PrP-sen would facilitate the accumulation of intracellular PrP-res to high enough levels to maintain both persistent infection in the mother cell and enable the transfer of organelles containing sufficient PrP-res to initiate infection in the daughter cell. Thus, we would suggest that efficient spread of infectivity requires not just the passive transfer of PrP-res from cell-to-cell but the concurrent initiation of conversion and amplification of PrP-res via cell surface, GPI anchored PrP-sen.
In vivo, several lines of evidence suggest that the spread of scrapie infectivity also requires de novo PrP-res formation in the recipient cell and not simply transfer of PrP-res from one cell to another. For example, when neurografts from PrP expressing mice were placed in the brains of PrP knockout mice and the mice were challenged intracranially with scrapie, the graft showed scrapie pathology, but the surrounding tissue did not.Citation30 Furthermore, PrP-res from the graft migrated to the host tissue demonstrating that simple transfer of PrP-res was not sufficient and that PrP-sen expression was required for the spread of scrapie pathology.Citation30 In fact, these mice did not develop scrapie pathology following peripheral infection even when peripheral lymphoid tissues were reconstituted with PrP-sen expressing cells.Citation31 Even though PrP-sen expressing cells were present in both the brain and spleen, in order for infectivity to spread from the lymphoreticular system to the central nervous system PrP-sen expression was also required in an intermediate tissue such as peripheral nerve.Citation31,Citation32 Given that PrP-res uptake and trafficking do not require PrP-sen, the most obvious explanation for the requirement of PrP-sen in contiguous tissues is that de novo PrP-res formation in naïve cells is necessary for (1) infectivity to move from cell to cell within a tissue and, (2) infectivity to move from tissue to tissue.
Another study demonstrated that peripheral expression of heterologous mouse PrP significantly increased the incubation time and actually prevented clinical disease in the majority of transgenic mice expressing hamster PrP in neurons of the brain.Citation33 Once again, if simple transfer and uptake of PrP-res were sufficient for spread, the presence of heterologous PrP molecules should not interfere because cellular uptake of PrP-res is independent of PrP-sen expression.Citation19,Citation21–Citation23 Clinical disease in these mice was likely prevented by the heterologous PrP molecule interfering with conversion of PrP-sen to PrP-res suggesting that prevention of de novo PrP-res formation inhibits spread of PrP-res and infectivity. These in vivo data, when combined with our recent in vitro data,Citation17 provide evidence to support the importance of cell surface, and by extension GPI-anchored, PrP in the spread of prion infection.
Our data demonstrate that the GPI anchor plays a role in the establishment of persistent scrapie infection in vitro. In our tissue culture system,Citation21 as well as others where spread of infectivity by cell to cell contact appears to be limited,Citation25,Citation34 the role of GPI anchored PrP-sen would be to amplify PrP-res to enable the efficient transfer of infectivity from mother to daughter cell. In cell systems where spread of prion infectivity may require cell to cell contact,Citation26,Citation27 we propose that the role of GPI anchored PrP-sen is to facilitate the spread of prion infection via a chain of conversion from cell-to-cell, a “domino” type spread of infection that has been previously hypothesized.Citation35,Citation36
In vivo, such a mechanism might explain why neuroinvasion does not necessarily require axonal transportCitation32,Citation37,Citation38 and can occur independently of the axonal neurofilament machinery.Citation39 It would likely vary with cell typeCitation27 and be most important in areas where infectivity is transferred from the periphery to the nervous system as well as in areas where cell division may be limited. It is also possible, if the location of PrP-res formation differs for different scrapie strains,Citation40 that the relative importance of a domino-like spread of infectivity in vivo would vary with the scrapie strain.
Of course, spread of infectivity via a “wave” of GPI anchored, PrP mediated conversion would not preclude the spread of infectivity by other intracellular means such as axonal transport (reviewed in ref. Citation41). Furthermore, spread of infectivity may still also occur extracellularly such as in the unique case of mice which express anchorless PrP-sen,Citation16 where our in vitro data would suggest that the cells themselves are not infected. In such a case, spread would require neither GPI anchored PrP-sen nor amplification of PrP-res in cells but would likely occur via other means such as bloodCitation41 or interstitial fluid flow.Citation42
Figures and Tables
Figure 1 Persistent infection of cells in vitro requires the expression of GPI-anchored cell surface PrP-sen. PrP knockout cells (CF10)Citation21 were transduced with 3F4 epitope tagged mouse PrP-sen (Mo3F4), 3F4 epitope tagged mouse PrP-sen without the GPI anchor (Mo3F4 GPI-), or Mo3F4 GPI-PrP-sen plus wild-type, GPI anchored mouse PrP-sen (MoPrP). The cells were then exposed to the mouse scrapie strain 22L and passaged. (A) The presence of 3F4 epitope tagged, cell surface mouse PrP-sen was assayed by FACS analysis of fixed, non-permeabilized cells. CF10 cells expressing the following mouse PrP-sen molecules were assayed: Mo3F4 (solid line); Mo3F4 GPI− (dashed line); Mo3F4 GPI− + MoPrP (dotted and dashed line); Mo3F4 GPI− + MoPrP infected with 22L scrapie (dotted line). Only cells expressing Mo3F4 PrP-sen were positive for cell surface, 3F4 epitope tagged PrP. (B) Persistent infection was analyzed by inoculating the cells intracranially into transgenic mice overexpressing MoPrP (Tga20 mice). Only cells expressing anchored mouse PrP-sen were susceptible to scrapie infection. Cells expressing anchorless mouse PrP-sen did not contain detectable infectivity in either the cells or the cellular supernatant (data not shown). Data in (B) are adapted from McNally 2009.Citation17
Acknowledgements
This research was supported by the Intramural Research Program of the NIH, National Institute of Allergy and Infectious Diseases (Project #1-Z01-AI000752-12).
References
- Stahl N, Borchelt DR, Prusiner SB. Differential release of cellular and scrapie prion proteins from cellular membranes by phosphatidylinositol-specific phospholipase C. Biochemistry 1990; 29:5405 - 5412
- Caughey B, Raymond GJ. The scrapie-associated form of PrP is made from a cell surface precursor that is both protease- and phospholipase-sensitive. J Biol Chem 1991; 266:18217 - 18223
- Borchelt DR, Taraboulos A, Prusiner SB. Evidence for synthesis of scrapie prion protein in the endocytic pathway. J Biol Chem 1992; 267:16188 - 16199
- Marijanovic Z, Caputo A, Campana V, Zurzolo C. Identification of an intracellular site of prion conversion. PLoS Pathog 2009; 5:1000426
- Kaneko K, Vey M, Scott M, Pilkuhn S, Cohen FE, Prusiner SB. COOH-terminal sequence of the cellular prion protein directs subcellular trafficking and controls conversion into the scrapie isoform. Proc Natl Acad Sci USA 1997; 94:2333 - 2338
- Taraboulos A, Scott M, Semenov A, Avraham D, Laszlo L, Prusiner SB. Cholesterol depletion and modification of COOH-terminal targeting sequence of the prion protein inhibit formation of the scrapie isoform. J Cell Biol 1995; 129:121 - 132
- Baron GS, Wehrly K, Dorward DW, Chesebro B, Caughey B. Conversion of raft associated prion protein to the protease-resistant state requires insertion of PrP-res (PrP(Sc)) into contiguous membranes. EMBO J 2002; 21:1031 - 1040
- Baron GS, Caughey B. Effect of glycosylphosphatidylinositol anchor-dependent and -independent prion protein association with model raft membranes on conversion to the protease-resistant Isoform. J Biol Chem 2003; 278:14883 - 14892
- Campana V, Sarnataro D, Zurzolo C. The highways and byways of prion protein trafficking. Trends Cell Biol 2005; 15:102 - 111
- Kocisko DA, Come JH, Priola SA, Chesebro B, Raymond GJ, Lansbury PT, et al. Cell-free formation of protease-resistant prion protein. Nature 1994; 370:471 - 474
- Deleault NR, Harris BT, Rees JR, Supattapone S. Formation of native prions from minimal components in vitro. Proc Natl Acad Sci USA 2007; 104:9741 - 9746
- Kirby L, Birkett CR, Rudyk H, Gilbert IH, Hope J. In vitro cell-free conversion of bacterial recombinant PrP to PrPres as a model for conversion. J Gen Virol 2003; 84:1013 - 1020
- Colby DW, Zhang Q, Wang S, Groth D, Legname G, Riesner D, et al. Prion detection by an amyloid seeding assay. Proc Natl Acad Sci USA 2007; 104:20914 - 20919
- Atarashi R, Moore RA, Sim VL, Hughson AG, Dorward DW, Onwubiko HA, et al. Ultrasensitive detection of scrapie prion protein using seeded conversion of recombinant prion protein. Nat Methods 2007; 4:645 - 650
- Legname G, Baskakov IV, Nguyen HO, Riesner D, Cohen FE, DeArmond SJ, et al. Synthetic mammalian prions. Science 2004; 305:673 - 676
- Chesebro B, Trifilo M, Race R, Meade-White K, Teng C, LaCasse R, et al. Anchorless prion protein results in infectious amyloid disease without clinical scrapie. Science 2005; 308:1435 - 1439
- McNally KL, Ward AE, Priola SA. Cells expressing anchorless prion protein are resistant to scrapie infection. J Virol 2009; 83:4469 - 4475
- Baron GS, Magalhaes AC, Prado MA, Caughey B. Mouse-adapted scrapie infection of SN56 cells: greater efficiency with microsome-associated versus purified PrP-res. J Virol 2006; 80:2106 - 2117
- Magalhaes AC, Baron GS, Lee KS, Steele-Mortimer O, Dorward D, Prado MA, et al. Uptake and neuritic transport of scrapie prion protein coincident with infection of neuronal cells. J Neurosci 2005; 25:5207 - 5216
- Gousset K, Schiff E, Langevin C, Marijanovic Z, Caputo A, Browman DT, et al. Prions hijack tunnelling nanotubes for intercellular spread. Nat Cell Biol 2009; 11:328 - 336
- Greil CS, Vorberg IM, Ward AE, Meade-White KD, Harris DA, Priola SA. Acute cellular uptake of abnormal prion protein is cell type and scrapie-strain independent. Virology 2008; 379:284 - 293
- Hijazi N, Kariv-Inbal Z, Gasset M, Gabizon R. PrPSc incorporation to cells requires endogenous glycosaminoglycan expression. J Biol Chem 2005; 280:17057 - 17061
- Paquet S, Daude N, Courageot MP, Chapuis J, Laude H, Vilette D. PrPc does not mediate internalization of PrPSc but is required at an early stage for de novo prion infection of Rov cells. J Virol 2007; 81:10786 - 10791
- Vorberg I, Raines A, Priola SA. Acute formation of protease-resistant prion protein does not always lead to persistent scrapie infection in vitro. J Biol Chem 2004; 279:29218 - 29225
- Ghaemmaghami S, Phuan PW, Perkins B, Ullman J, May BC, Cohen FE, et al. Cell division modulates prion accumulation in cultured cells. Proc Natl Acad Sci USA 2007; 104:17971 - 17976
- Kanu N, Imokawa Y, Drechsel DN, Williamson RA, Birkett CR, Bostock CJ, et al. Transfer of scrapie prion infectivity by cell contact in culture. Curr Biol 2002; 12:523 - 530
- Paquet S, Langevin C, Chapuis J, Jackson GS, Laude H, Vilette D. Efficient dissemination of prions through preferential transmission to nearby cells. J Gen Virol 2007; 88:706 - 713
- Fevrier B, Vilette D, Archer F, Loew D, Faigle W, Vidal M, et al. Cells release prions in association with exosomes. Proc Natl Acad Sci USA 2004; 101:9683 - 9688
- Mishra RS, Basu S, Gu Y, Luo X, Zou WQ, Mishra R, et al. Protease-resistant human prion protein and ferritin are cotransported across Caco-2 epithelial cells: implications for species barrier in prion uptake from the intestine. J Neurosci 2004; 24:11280 - 11290
- Brandner S, Isenmann S, Raeber A, Fischer M, Sailer A, Kobayashi Y, et al. Normal host prion protein necessary for scrapie-induced neurotoxicity. Nature 1996; 379:339 - 343
- Blattler T, Brandner S, Raeber AJ, Klein MA, Voigtlander T, Weissmann C, et al. PrP-expressing tissue required for transfer of scrapie infectivity from spleen to brain. Nature 1997; 389:69 - 73
- Glatzel M, Aguzzi A. PrP(C) expression in the peripheral nervous system is a determinant of prion neuroinvasion. J Gen Virol 2000; 81:2813 - 2821
- Race R, Oldstone M, Chesebro B. Entry versus blockade of brain infection following oral or intraperitoneal scrapie administration: Role of prion protein expression in peripheral nerves and spleen. J Virol 2000; 74:828 - 833
- Vorberg I, Raines A, Story B, Priola SA. Susceptibility of common fibroblast cell lines to transmissible spongiform encephalopathy agents. J Infect Dis 2004; 189:431 - 439
- Glatzel M, Aguzzi A. Peripheral pathogenesis of prion diseases. Microbes Infect 2000; 2:613 - 619
- Follet J, Lemaire-Vieille C, Blanquet-Grossard F, Podevin-Dimster V, Lehmann S, Chauvin JP, et al. PrP expression and replication by Schwann cells: implications in prion spreading. J Virol 2002; 76:2434 - 2439
- Groschup MH, Beekes M, McBride PA, Hardt M, Hainfellner JA, Budka H. Deposition of disease-associated prion protein involves the peripheral nervous system in experimental scrapie. Acta Neuropathol 1999; 98:453 - 457
- Hainfellner JA, Budka H. Disease associated prion protein may deposit in the peripheral nervous system in human transmissible spongiform encephalopathies. Acta Neuropathol 1999; 98:458 - 460
- Kratzel C, Kruger D, Beekes M. Prion propagation in a nerve conduit model containing segments devoid of axons. J Gen Virol 2007; 88:3479 - 3485
- Vorberg I, Priola SA. Molecular basis of scrapie strain glycoform variation. J Biol Chem 2002; 277:36775 - 36781
- Beekes M, McBride PA. The spread of prions through the body in naturally acquired transmissible spongiform encephalopathies. FEBS J 2007; 274:588 - 605
- Weller RO. Pathology of cerebrospinal fluid and interstitial fluid of the CNS: significance for Alzheimer disease, prion disorders and multiple sclerosis. J Neuropathol Exp Neurol 1998; 57:885 - 894