
Abstract
Increasing evidence suggests that proteins exhibiting “prion-like” behavior cause distinct neurodegenerative diseases, including inherited, sporadic and acquired types. The conversion of cellular prion protein (PrPC) to its infectious protease resistant counterpart (PrPRes) is the essential feature of prion diseases. However, PrPC also performs important functions in transmembrane signaling, especially in neurodegenerative processes. Beta-amyloid (Aβ) synaptotoxicity and cognitive dysfunction in mouse models of Alzheimer disease are mediated by a PrPC-dependent pathway. Here we review how this pathway converges with proinflammatory cytokine signaling to activate membrane NADPH oxidase (NOX) and generate reactive oxygen species (ROS) leading to dynamic remodeling of the actin cytoskeleton. The NOX signaling pathway may also be integrated with those of other transmembrane receptors clustered in PrPC-enriched membrane domains. Such a signal convergence along the PrPC-NOX axis could explain the relevance of PrPC in a broad spectrum of neurodegenerative disorders, including neuroinflammatory-mediated alterations in synaptic function following traumatic brain injury. PrPC overexpression alone activates NOX and generates a local increase in ROS that initiates cofilin activation and formation of cofilin-saturated actin bundles (rods). Rods sequester cofilin from synaptic regions where it is required for plasticity associated with learning and memory. Rods can also interrupt vesicular transport by occluding the neurite within which they form. Through either or both mechanisms, rods may directly mediate the synaptic dysfunction that accompanies various neurodegenerative disorders.
Introduction
The cellular prion protein (PrPC) is expressed in many cell types but is particularly enriched in neurons. Here we review evidence that PrPC has a role in transmembrane signaling in neurons. We will discuss a possible mechanism by which diverse extracellular ligands, such as amyloid-β and proinflammatory cytokines, utilize PrPC to induce an identical alteration in the actin cytoskeleton that affects synaptic function.
The encoded PrPC contains an N-terminal signal peptide removed after transport across the endoplasmic reticulum and a C-terminal peptide that is removed when the protein is transferred to its glycosylphosphatidylinositiol (GPI) lipid that keeps it anchored in the outer leaflet of the plasma membrane (). Near the N-terminus of the processed protein are 4 to 5 octapeptide repeats that contain up to 5 copper binding sites.Citation1 Copper binding induces significant conformational changes to PrPC, which may have important implications in prion formation and in modulating PrPC interactions with other membrane proteins.Citation2 Neuronal, GPI-anchored PrPC serves as a relay for transmissible spongiform encephalopathy (prion disease)Citation3 induced by the β-sheet-containing form of the prion protein, referred to as PrPSc (scrapie) or PrPRes (protease resistant). The C-terminal region of PrPC contains a sphingolipid-binding domain and 3 α helices that undergo a conformational transformation to β-sheet when the PrPC interacts with an appropriate PrPRes. The converted PrPRes self-assembles into fibrils and insoluble aggregates that accumulate in brain. Although PrPC is highly conserved across mammalian species, only a few amino acid differences, of which many may be conservative, are sufficient to provide a species barrier for prion disease transmission.
Figure 1. Schematic of structural domains and interactions of PrPC. PrPC contains an N-terminal signal sequence (1–22) and 2 charged clusters (CC1/2) on either side of 5 copper-binding octapeptide repeats (OR). Two principal binding sites have been identified for Aβ (23–50 and 92–110) whereas caveolin-1 has been shown to interact with ORs, where it may serve to link cellular prion to the activation of fyn tyrosine-kinase. The C-terminal domain contains 3 α helices, 2 of which are stabilized by a disulfide bond, while a third (α1) unfolds and is converted to the PrPRes conformation. A sphingolipid binding domain is located between the 2 cysteine residues (179 and 214). GPI attachment at amino acid 231 anchors PrPC to the extracellular leaflet of the plasma membrane upon elimination of the C-terminal peptide (232–254).
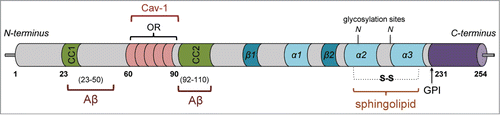
PrPC contains 2 potentially glycosylated asparagine residues,Citation2 undergoes 3 different proteolytic cleavages, and thus, exists as multiple post-translationally modified isoforms whose distribution and relevance may vary based on distinct brain region or cell type. This concept of prion speciation might account for its ability to affect specific neuronal populations and thus play a role in a spectrum of neurodegenerative disorders beyond those induced by infectious prions. These include Alzheimer disease (AD), Parkinson disease, Huntington disease, other frontotemporal dementias such as chronic traumatic encephalopathy, as well as traumatic brain injury (TBI).Citation4,5 Although PrPC is widely expressed in neurons, there are certain neuronal populations and neurite populations in which its expression is below detection.Citation6 In addition and of relevance to implications on synaptic function, PrPC was found highly enriched in post-synaptic densities.Citation7
Knockout of the PrPC-encoding gene Prnp in mice results in viable animals that develop normally but which are resistant to prion infection.Citation2 Although the animals show increased vulnerability to various types of stress, specific roles for the PrPC in normal function have been difficult to identify. However, PrPC was suggested to be a protein involved in signal transduction many years agoCitation8 and recent work supports a role for PrPC as a receptor or co-receptor to mediate signal transduction processes, some of which may be especially important in the neurodegenerative cascade leading to loss of synaptic function.
PrPC is a component of distinct membrane microdomains
Transmembrane and membrane associated proteins have lipid interacting domains of various types that are important for their conformation and function. Many membrane proteins have transmembrane segments that are accommodated better within slightly thicker membrane microdomains, often referred to as lipid rafts, containing phosphatidylinositols and cholesterol and in which sphingolipids are concentrated in the exoplasmic face of the plasma membrane. Although their exact character is debated, lipid rafts are spatially and temporally dynamic and play a vital role in many cellular signaling pathways, including those of cytokines, growth factors, and hormones.Citation9 GPI-linked proteins, such as PrPC, are also concentrated in these domains. Specific hydrogen bonding between cholesterol and sphingomyelin appears critical for the formation and stability of lipid rafts. The biophysical properties of the lipid raft environment affect the recruitment, conformation, interaction, and physiological function of raft-associated proteins. Several ligand-gated ion channels and G-protein coupled receptors contain specific binding sites, either in their transmembrane helices or extracellular loop structures, for cholesterol and sphingolipids, the binding of which allows conformational changes necessary for function.Citation10 Subtle changes in lipid species alter membrane architecture and impose constraints on lipid-protein and/or protein-protein interactions influencing the function of membrane proteins.
Levels of raft-associated lipids such as sphingomyelin, ceramides, and cholesterol significantly increase with age,Citation11 suggesting that lipid raft densities and associated signaling process are likely subjected to age-dependent alterations that could contribute to the initiation and/or progression of AD,Citation9 as could dietary factors that affect lipid accumulation and metabolism and alterations in membrane lipids resulting from oxidative stress (e.g. lipid peroxidation).Citation12 Although the lipid raft microenvironment is essential for the conversion of PrPC to the neurotoxic PrPRes,Citation13 perhaps through the GM1 ganglioside binding epitope in the C-terminal portion implicated in conferring structural changes to the prion protein,Citation14 the interactions of PrPC with both lipid and protein components within the membrane microdomains also are likely to influence the signaling output of the system.
PrPC links Aβ and proinflammatory cytokines to cognitive impairment
GPI-anchored PrPC serves as a receptor for Aβ oligomersCitation2,7 () and is required for the synaptotoxicity and cognitive deficits of Aβ oligomers in mice.Citation2,7,15 Cognitive decline in human subjects appears to depend more on the length (Aβ1–42 vs shorter forms) and post-production alterations of Aβ (formation of dimers/oligomers) than simply the total amount produced.Citation16 SDS-stable dimers are a major form of soluble Aβ extracted from most AD brains and may be the essential building block of the most synaptotoxic Aβ species that can induce dendritic spine loss and AD progression.Citation16,17
Synapse loss can be detected in amyloid precursor protein (APP)swe/presenilin 1 (PS1)ΔE9 mice by staining for synaptic marker proteins, and the loss of synaptic markers is dependent on the presence of PrPC.Citation7 Treatment of aged APPswe/PS1ΔE9 mice with anti-PrPC antibodies allows a recovery of synaptic markers in the dentate gyrus of the hippocampus, a brain region associated with learning and memory.Citation18
Neuroinflammation occurs in TBI and many other neurodegenerative disorders. Proinflammatory cytokines, including tumor necrosis factor α (TNFα) and interleukin (IL)-1β, are increased in mild TBI and modulate deficits in motor coordination.Citation19 Although the molecular mechanism by which the proinflammatory cytokines mediate the decline in cognitive function associated with the neuroinflammatory responses in mild TBI is incompletely understood, neuronal receptors for these cytokines are concentrated in lipid raft domains, where they enhance redox-signaling events.Citation20,21
PrPC dependency for cofilin-actin rod formation induced by Aβ and proinflammatory cytokines
Dynamic remodeling of the actin cytoskeleton into rod-like neuritic inclusions may be a final common mechanism for synaptic dysfunction in many neurodegenerative disorders. Activation (dephosphorylation) of actin-depolymerizing factor (ADF)/cofilin, a family of proteins that enhances the severing and turnover of actin filaments, initiates the formation of short, stable ADF/cofilin-saturated actin filaments, which under oxidative conditions can bundle to form rod-like inclusions (rods) within neurites ().Citation22 Cofilin is 5–10 fold more abundant in mammalian neurons than ADF, thus the rods are called cofilin-actin rods. Rod-like neuritic inclusions have been observed in human AD brain and in mouse models of AD.Citation23,24 Rods can grow to occlude neurites, blocking vesicular transport necessary for synaptic function.Citation23,25,26 Specifically, formation of rods results in a global loss of trafficking of amyloid precursor protein (APP)-containing vesiclesCitation27 that normally traffic both anterogradely to the plasma membrane and retrogradely for lysosomal degradation.Citation28 Rods may also mediate synaptic dysfunction by sequestering ADF/cofilin. In dendritic spines, cofilin functions in many aspects of receptor insertion and spine dynamics associated with memory and learning, whereas ADF/cofilin together appear to function in presynaptiC-terminals.Citation29
Figure 2:. Schematic model of PrPC-mediated membrane signaling complex for Aβ and proinflammatory cytokine-induced cofilin-actin rod formation. This proposed hypothetical model links Aβd/t and the proinflammatory cytokine TNFα to cofilin-actin rod formation in membrane raft domains in which PrPC is highly concentrated. Overexpression of PrPC alone is sufficient to induce rods in a NOX-dependent manner, suggesting that enlargement of membrane signaling domains from which resultant ROS generation exceeds a rod-inducing threshold is regulated by PrPC levels. This inducible oxidizing environment both maintains dephosphorylation-dependent activation of cofilin and initiates intermolecular disulfide bond formation in dephosphorylated cofilin to permit bundling of cofilin-saturated actin filaments. The dynamics of rod dissolution and reversal upon addition of several different NOX inhibitors further suggests that rods may be transient in nature until such time as the stimulus for their formation maintains ROS above the threshold required for rod maintenance.
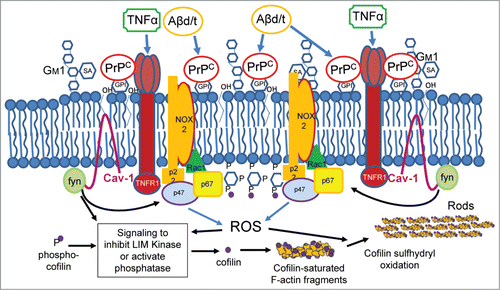
Rod formation is triggered in cultured neurons by Aβ oligomers, but especially by those forms most associated with AD progression, soluble Aβ dimer/trimer (Aβd/t).Citation30,31 Rods are also induced by several proinflammatory cytokines in the same neuronal subpopulation that responds to Aβd/t, and both inducers require the expression of PrPC for rod signaling.Citation31 Formation of rods downstream from PrPC is dependent on the activation of NADPH oxidase (NOX) for production of reactive oxygen species (ROS), which is required for oxidation of cofilin sulfhydryls to form an intermolecular disulfide bond.Citation22,31 Rods induced by Aβ or proinflammatory cytokines are reversible and their stability appears to depend upon the continuous production of ROS above a threshold level to maintain them. Addition of NOX inhibitors or washout of the inducer reverses rod formation with a similar half-life of about 35 min.Citation31
Previous work has demonstrated that several different Aβ-binding proteins are able to mediate cognitive decline,Citation2 including at least one (mGluR5) for which an association with PrPC has been established.Citation7 Each of the cytokines shown to induce rods in the same population of neurons as Aβ has its own unique receptor and all of them use a PrPC-dependent pathway for rod formation that involves NOX activation and ROS production. NOX activity in human brain is inversely correlated with cognitionCitation30 as its inhibition had beneficial effects on various central nervous system pathologies as well as inhibition and reversal of PrPC-induced rods in cultured neurons.Citation31 Thus, rod formation represents a common mechanism by which a multitude of membrane proteins that bind the promiscuous Aβ could work through a PrPC-dependent signaling pathway to affect synapse plasticity and ultimately cognitive function.
PrPC signaling in synaptic loss
Surprisingly, the overexpression of PrPC alone is sufficient to induce cofilin-actin rods.Citation31 At low levels of PrPC-overexpression, rod formation is enhanced by the presence of a stressor such as Aβd/t or TNFα, but at higher levels of expression, no additional rod formation occurs with stressor addition. Rods induced by PrPC-overexpression are reversed by inhibiting NOX activity, suggesting the clustering of GPI-anchored PrPC into membrane domains independently recruits and activates NOX for ROS production. This idea is reinforced by studies that show the clustering of GPI-linked PrPC alone alters the membrane lipid profile and results in density-dependent synaptic damage.Citation32 Both Aβ and TNFα activate neutral sphingomyelinase generating ceramide and significantly changing the lipid raft environment.Citation21,33 Such changes could contribute to the proposed signaling pathway by altering interactions between raft components.
NOX-mediated ROS production occurs downstream of fyn, a non-receptor tyrosine kinase that is activated in a PrPC-dependent manner by Aβ7 or by antibody cross-linking.Citation2 Fyn activation occurs mainly in neurites where a conformational change in PrPC results in its octapeptide repeat region interacting directly with the hairpin-like membrane-embedded protein caveolin-1.Citation2,34 Both of these interactions result in caveolin-1 and fyn phosphorylation. Thus, it is easy to visualize how alterations in metal ion binding to the octapeptide repeats or in the lipid composition of raft domains could influence PrPC signaling. Fyn activation through PrPC may mediate both neural cell adhesion molecule-dependent neurite outgrowth and synaptic dysfunction.Citation2 The downstream effects of PrPC-fyn signaling, which includes NOX activation by phosphorylation and enhanced recruitment of cytoplasmic NOX subunits, are enhanced by Aβ, which clusters and crosslinks PrPC at the neuronal cell membrane.Citation2,32 Aβ-dependent PrPC-fyn signaling has been tied to the overactivation of metabotropic mGluR5 receptors as well as the NMDA receptorCitation2,7 and may utilize a Ca2+-dependent mechanism for synaptic dysregulation.
Although the specific pathways utilized for Aβ or proinflammatory cytokine activation (dephosphorylation) of cofilin have not been conclusively established, several have been characterized in neurons and may be utilized. The cofilin phosphatase slingshot (SSH) is activated by ROS via the oxidation-dependent removal of 14-3-3 isoform which is inhibitory to SSH activation.Citation25 SSH can also be directly activated by dephosphorylation via the Ca2+-dependent phosphatase calcineurin. Alternatively, the cofilin phosphatase chronophin is activated by release from Hsp90 which occurs upon localized ATP depletion.Citation35 In addition, there are potential cofilin-activating pathways downstream of fyn.
About 20% of cultured hippocampal neurons globally treated with Aβd/t or proinflammatory cytokines form rods and these rods form in a subset of neurites in which cofilin activation occurs. More than 40% of neurons will form rods in response to the overexpression of PrPC in the absence of any additional treatment. Although there is a small but significant (P < 0.001) increase in the number of rods per cell body (from 3.5 in Aβ-treated neurons to 3.9 in neurons overexpressing PrPC), the number of neurites in which rods form does not significantly increase (average of 1.54 neurites per neuron with Aβ treatment versus 1.59 in PrPC overexpressing neurons). However, in >80% of neurons exposed to excitotoxic levels of glutamate or that are energy-depleted (i.e. mitochondrial inhibition), rods form within minutes and are present in most neurites. This rapid pathway for rod formation does not require the presence of PrPC or NOX activity.Citation31 The above findings suggest that the components of rods are not limiting but that for PrPC-dependent rod formation some other factor downstream of PrPC restricts the signaling pathway to specific neurites in a subset of neurons. The nature of this factor is unknown but it could be caveolin-1, whose presence in neurons was debated for many years because it went undetected by immunostaining.
PrPC-fyn signaling also promotes tau hyperphosphorylation in response to oligomeric Aβ.Citation2 The hyperphosphorylation of tau follows its release from microtubules and enhances its assembly into paired helical filaments (PHFs). The PHFs aggregate to form neuropil threads and neurofibrillary tangles, the latter of which, along with amyloid plaques, are considered the pathological hallmarks of AD, but do not always correlate well with cognitive status.Citation36,37 However, cofilin-actin rods can disrupt microtubules, causing the release of tau.Citation23 Because rods are reversed upon the removal of the inducing agent or a decline in ROS below the threshold required for rod maintenance, rods may initiate many changes in neurons but not be present throughout the degenerative cascade.Citation27,31
Concluding Remarks
Cofilin-actin rod formation provides a common end point for signaling from multiple receptors that interact with PrPC to stimulate ROS production from NOX. That overexpression of PrPC alone initiates this pathway suggests that the role of Aβ and other ligands (e.g., proinflammatory cytokines) is to enhance the interactions of membrane raft components for NOX activation (). Since this proposed signaling pathway occurs in the lipid raft environment, distinct lipid species could influence either positively or negatively the conformational changes of proteins involved.
AD is a disease of neuronal connectivity with synapse loss its earliest feature. Given that sporadic AD is the most common form and is likely initiated by a diversity of factors, it would be naïve to think that rods explain all of the synaptic dysfunction that arises. Nevertheless, the rapidity with which rods form, their ability to reverse upon reduction in ROS, and their ability to become persistent with continued elevation of ROS suggest that rods could be an important effector of neurodegenerative signaling and synapse dysfunction. The requirement for a ROS threshold to maintain rods might explain why a decrease in a number of different raft-associated Aβ binding partners has beneficial effects on cognitive function in mouse models.
Acknowledgment
We thank Laurie Minamide, Alisa Shaw and Dr. Mark Zabel for comments and critical reading of the manuscript.
Funding
Supported in part by a grant from National Institutes of Health (AG044812 to JRB).
References
- Jackson GS, Murray I, Hosszu LL, Gibbs N, Waltho JP, Clarke AR, Collinge J. Location and properties of metal-binding sites on the human prion protein. Proc Natl Acad Sci 2001; 98:8531-5; PMID:11438695; http://dx.doi.org/10.1073/pnas.151038498
- Hernandez-Rapp J, Martin-Lannerée S, Hirsch TZ, Launay JM, Mouillet-Richard S. Hijacking PrPC-dependent signal transduction: when prions impair Aβ clearance. Front Aging Neurosci 2014; 6:25-30; PMID:24592237; http://dx.doi.org/10.3389/fnagi.2014.00025
- Ermonval M, Mouillet-Richard S, Codogno P, Kellermann O, Botti J. Evolving views in prion glycosylation: functional and pathological implications. Biochimie 2003; 85:33-45; PMID:12765773; http://dx.doi.org/10.1016/S0300-9084(03)00040-3
- Prusiner SB. Biology and genetics of prions causing neurodegeneration. Annu Rev Genet 2013; 47:601-23; PMID:24274755; http://dx.doi.org/10.1146/annurev-genet-110711-155524
- Renner M, Melki R. Protein aggregation and prionopathies. Pathol Biol 2014; S0369-8114: 33-9
- Barmada S, Piccardo P, Yamaguchi K, Ghetti B, Harris DA. GFP-tagged prion protein is correctly localized and functionally active in the brains of transgenic mice. Neurobiol Dis 2004; 16:527-37; PMID:15262264; http://dx.doi.org/10.1016/j.nbd.2004.05.005
- Um JW, Strittmatter SM. Amyloid-β induced signaling by cellular prion protein and Fyn kinase in Alzheimer disease. Prion 2013; 7:37-41; PMID:22987042; http://dx.doi.org/10.4161/pri.22212
- Mouillet-Richard S, Ermonval M, Chebassier C, Laplanche JL, Lehmann S, Launay JM, Kellermann O. Signal transduction through prion protein. Science 2000; 289:1925-8; PMID:10988071; http://dx.doi.org/10.1126/science.289.5486.1925
- Haughey NJ, Veera VR, Mihyun B, Mattson MP. Roles for dysfunctional sphingolipid metabolism in Alzheimer's disease neuropathogenesis. Biochim Biophys Acta 2010; 1801:878-86; PMID:20452460; http://dx.doi.org/10.1016/j.bbalip.2010.05.003
- Fantini J, Barrantes FJ. Sphingolipidcholesterol regulation of neurotransmitter receptor conformation and function. Biochim Biophys Acta 2009; 1788:2345-61; PMID: 19733149; http://dx.doi.org/10.1016/j.bbamem.2009.08.016
- Michel V., Bakovic M. Lipid rafts in health and disease. Bio Cell 2007; 99:129-40; PMID:17064251; http://dx.doi.org/10.1042/BC20060051
- Sultana R, Perluigi M, Butterfield DA. Lipid peroxidation triggers neurodegeneration: a redox proteomics view into Alzheimer disease brain. Free Radic Biol Med 2013; 62:157-69; PMID:23044265; http://dx.doi.org/10.1016/j.freeradbiomed.2012.09.027
- Taylor DR, Hooper NM. The prion protein and lipid rafts. Mol Memb Biol 2006; 31:89-99; PMID:16611584; http://dx.doi.org/10.1080/09687860500449994
- Sanghera N, Correia BEFS, Correia JRS, Ludwig C, Agarwal S, Nakamura HK, Kuwata K, Samain E, Gill AC, Bonev BB, et al. Deciphering the molecular details for the binding of the prion protein to main ganglioside GM1 of neuronal membranes. Chem Biol 2011; 18:1422-31; PMID:22118676; http://dx.doi.org/10.1016/j.chembiol.2011.08.016
- Pradines E, Hernandez-Rapp J, Villa-Diaz A, Dakowski C, Ardila-Osorio H, Haik S, Schneider B, Launay LM, Kellerman O, Torres JM, et al. Pathogenic prions deviate PrPC signaling in neuronal cells and impair A-beta clearance. Cell Death Dis 2013; 4:e456; PMID:23303130; http://dx.doi.org/10.1038/cddis.2012.195
- Masters CL, Selkoe DJ. Biochemistry of Amyloid β-protein and amyloid deposits in Alzheimer disease. Cold Spring Harb Perspect Med 2012; 2:a006262; PMID: 22675658; http://dx.doi.org/10.1101/cshperspect.a006262
- McDonald JM, Cairns NJ, Taylor-Reinwald L, Holtzman D, Walsh DM. The levels of water-soluble and triton-soluble Aβ are increased in Alzheimer's disease brain. Brain Res 2012; 1450:138-47; PMID:22440675; http://dx.doi.org/10.1016/j.brainres.2012.02.041
- Chung E, Ji Y, Sun Y, Kascsak RJ, Kascsak RB, Mehta PD, Strittmatter SM, Wisniewski T. Anti-PrPC monoclonal antibody infusion as a novel treatment for cognitive deficits in an Alzheimer's disease model mouse. BMC Neurosci 2010; 11:130-40; PMID:20946660; http://dx.doi.org/10.1186/1471-2202-11-130
- Lin Y, Wen L. Inflammatory response following diffuse axonal injury. Int J Med Sci 2013; 10:515-21; PMID:23532682; http://dx.doi.org/10.7150/ijms.5423
- Yang B, Rizzo V. TNF-alpha potentiates protein-tyrosine nitration through activation of NADPH oxidase and eNOS localized in membrane rafts and caveolae of bovine aortic endothelial cells. Am J Physiol Heart Circ Physiol 2007; 292:H954-62; PMID:17028163; http://dx.doi.org/10.1152/ajpheart.00758.2006
- Barth BM, Gustafson SJ, Hankins JL, Kaiser JM, Haakenson JK, Kester M, Kuhn TB. Ceramide kinase regulates TNFα-stimulated NADPH oxidase activity and eicosanoid biosynthesis in neuroblastoma cells. Cell Signal 2012; 24:1126-33; PMID:22230689; http://dx.doi.org/10.1016/j.cellsig.2011.12.020
- Bernstein BW, Shaw AE, Minamide LS, Pak CW, Bamburg JR. Incorporation of cofilin into rods depends on disulfide intermolecular bonds: implications for actin regulation in neurodegenerative disease. J Neurosci 2012; 32:6670-81; PMID:22573689; http://dx.doi.org/10.1523/JNEUROSCI.6020-11.2012
- Minamide LS, Striegl AM, Boyle JA, Meberg PF, Bamburg JR. Neurodegenerative stimuli induce persistent ADFcofilin-actin rods that disrupt distal neurite function. Nat Cell Biol 2000; 2: 628-36; PMID:10980704; http://dx.doi.org/10.1038/35023579
- Maloney MT, Minamide LS, Kinley AW, Boyle JA, Bamburg JR. Beta-secretase-cleaved amyloid precursor proteins accumulates at actin inclusions induced in neurons by stress or amyloid beta: a feedforward mechanism for Alzheimer's disease. J Neurosci 2005; 25: 11313-21; PMID:16339026; http://dx.doi.org/10.1523/JNEUROSCI.3711-05.2005
- Schönhofen P, Marengo de Medeiros L, Chatain CP, Bristot IJ, Klamt F. Cofilinactin rod formation by dysregulation of cofilin-1 activity as a central step in neurodegeneration. Mini Rev Med Chem 2014; 14:393-400; PMID: 24813767; http://dx.doi.org/10.2174/1389557514666140506161458
- Cichon J, Sun C, Chen B, Jiang M, Cheng XA, Sun Y, Wang Y, Chen G. Cofilin aggregation blocks intracellular trafficking and induces synaptic loss in hippocampal neurons. J Biol Chem 2012; 287:3919-29; PMID:22184127; http://dx.doi.org/10.1074/jbc.M111.301911
- Mi J, Shaw AE, Pak CW, Walsh KP, Minamide LS, Bernstein BW, Kuhn TB, Bamburg JR. A genetically encoded reporter for real-time imaging of cofilin-actin rods in living neurons. PLoS One 2013; 8:e83609; PMID:24391794; http://dx.doi.org/10.1371/journal.pone.0083609
- Thinakaran G, Koo EH. Amyloid precursor protein trafficking, processing, and function. J Biol Chem 2008; 283:29615-9; PMID:18650430; http://dx.doi.org/10.1074/jbc.R800019200
- Wolf M, Zimmermann AM, Gorlich A, Gurniak CB, Sassoe-Pognetto M, Friauf E, Witke W, Rust MB. ADF/cofilin controls synaptic actin dynamics and regulates vesicle mobilization and exocytosis. Cereb Cortex 2014; PMID:24770705
- Davis RC, Marsden IT, Maloney MT, Minamide LS, Podlisny M, Selkoe DJ, Bamburg JR. Amyloid beta dimerstrimers potently induce cofilin-actin rods that are inhibited by maintaining cofilin phosphorylation. Mol Neurodegen 2011; 6:10; http://dx.doi.org/10.1186/1750-1326-6-10
- Walsh KP, Minamide LS, Kane SJ, Shaw AE, Brown DR, Pulford B, Zabel MD, Lambeth JD, Kuhn TB, Bamburg JR. Amyloid-β and proinflammatory cytokines utilize a prion protein-dependent pathway to activate NADPH oxidase and induce cofilin-actin rods in hippocampal neurons. PLoS One 2014; 9:e95995; PMID:24760020; http://dx.doi.org/10.1371/journal.pone.0095995
- Bate C, Williams A. Clustering of sialylated glycosylphosphatidylinositol anchors mediates PrP-induced activation of cytoplasmic phospholipase A2 and synapse damage. Prion 2012; 6:350-3; PMID:22895089; http://dx.doi.org/10.4161/pri.21751
- Clarke CJ, Wu BX, Hannun YA. The neutral sphingomyelinase family: identifying biochemical connections. Adv Enzyme Regul 2011; 51:51-8; PMID:21035485; http://dx.doi.org/10.1016/j.advenzreg.2010.09.016
- Shi Q, Jing YY, Wang SB, Chen C, Sun H, Xu Y, Gao C, Zhang J, Tian C, Guo Y, et al. PrP octarepeats region determined the interaction with caveolin-1 and phosphorylation of caveolin-1 and fyn. Med Microbiol Immunol 2013; 202:215-27; PMID:23283514; http://dx.doi.org/10.1007/s00430-012-0284-8
- Huang TY, Minamide LS, Bamburg JR, Bokoch GM. Chronophin mediates an ATP-sensing mechanism for cofilin dephosphorylation and neuronal cofilin-actin rod formation. Dev Cell 2008; 15:691-703; PMID:19000834; http://dx.doi.org/10.1016/j.devcel.2008.09.017
- Hernandez F, Avila J. Tauopathies. Cell Mol Life Sci 2007; 64:2219-33; PMID:17604998; http://dx.doi.org/10.1007/s00018-007-7220-x
- Scheff SW, Neltner JH, Nelson PT. Is synaptic loss a unique hallmark of Alzheimer's disease? Biochem Pharmacol 2014; 88:517-28; PMID:24412275; http://dx.doi.org/10.1016/j.bcp.2013.12.028
- Schneider B, Mutel V, Pietri M, Ermoval M, Mouillet-Richard S, Kellermann O. NADPH oxidase and extracellular regulated kinases 12 are targets of prion protein signaling in neuronal and nonneuronal cells. Proc Natl Acad Sci 2003; 100:13326-31; PMID:14597699; http://dx.doi.org/10.1073/pnas.2235648100
- Ansari MA, Scheff SW. NADPH-oxidase activation and cognition in Alzheimer disease progression. Free Radic Biol Med 2011; 51:171-8; PMID:21457777; http://dx.doi.org/10.1016/j.freeradbiomed.2011.03.025
- Head BP, Patel HH, Insel PA. Interaction of membranelipid rafts with the cytoskeleton: Impact on signaling and function. Biochim Biophys Acta 2014; 1838:532-45; PMID:23899502; http://dx.doi.org/10.1016/j.bbamem.2013.07.018