Abstract
The fruit fly Drosophila melanogaster has been a favored tool for genetic studies for over 100 years and has become an excellent model system to study development, signal transduction, cell biology, immunity, and behavior. The relevance of Drosophila to humans is perhaps best illustrated by the fact that more than 75% of the genes identified in human diseases have counterparts in Drosophila. During the last decade, many fly models of neurodegenerative disorders have contributed to the identification of novel pathways mediating pathogenesis. However, the development of prion disease models in flies has been remarkably challenging. We recently reported a Drosophila model of sporadic prion pathology that shares relevant features with the typical disease in mammals. This new model provides the basis to explore relevant aspects of the biology of the prion protein, such as uncovering the genetic mechanisms regulating prion protein misfolding and prion-induced neurodegeneration, in a dynamic, genetically tractable in vivo system.
Fruit Flies in the Genomics Age
Drosophila has played a key role in understanding many basic biology questions, particularly in the fields of genetics and development. In recent years, the fruit fly Drosophila melanogaster has emerged as an attractive model for studying human biology and disease, including development, tumorigenesis, degeneration, aging, innate immunity and complex behaviors such as addiction, learning and memory, and sleep. The reasons for working with fruit flies are multiple, the most important being the basic conservation of metabolic pathways, cellular organization and genetic constitution, as recently confirmed with the completion of the DrosophilaCitation1 and human genomes.Citation2,Citation3 However, more practical reasons have made Drosophila a favorite for genetic studies: a very compact genome distributed in three main chromosomes (and a tiny fourth chromosome) and a short life cycle (ten days). These attributes made possible the amazing genetic discoveries of Morgan, Mueller and Sturtevant in the early decades of the 20th century. Nowadays fruit flies are widely appreciated for many other experimental advantages, including well-characterized development, easy manipulation of all developmental stages, access to large collections of mutant strains, simple transgenesis and complete genome sequence.Citation4
Drosophila researchers still maintain an advantage over other animal models thanks to the availability of multiple strategies for controlling gene expression, including the acquisition of the flexible UAS/GAL4 system.Citation5 This dual expression system imported from yeast consists of two independent strains, one carrying a transgene of interest under the control of several copies of the UAS (Upstream Activating Sequence) promoter sequence, and a second strain expressing its transcriptional activator, the transcription factor Gal4. The transgene of interest is inserted into a freely available vector (pUAST) downstream of the UAS followed by injection in syncytial embryonic stages by well-established techniques.Citation6 Embryonic injections have become routine laboratory practice in the last couple of decades, but efficient and cheap ($150–200) commercial services have rendered laboratory injections obsolete. An important advantage of this dual regulatory system is that once several independent strains (5–20) are isolated, the transgenes are silent (not expressed) because flies lack Gal4 activity. Thus, strains can be easily stored regardless of the potential deleterious effects of the transgene. To induce transgene expression, the UAS-transgene strains are combined (crossed) with strains expressing Gal4 under the control of endogenous or engineered promoters. Hundreds of Gal4 strains have been generated by the Drosophila community and made available to other investigators, constituting an unparallel resource for research. These strains express Gal4 in an almost unlimited array of expression patterns with unique localization (tissue, territory, cell), levels (high, low) and timing (early, late, pre-, post-mitotic) to produce almost any desired combination.
As good as the UAS/Gal4 regulatory system has been for the spatial regulation of transgenes, this system presents an important shortcoming: UAS/Gal4 does not allow for efficient temporal regulation. To deal with this limitation, several regulatory systems have been developed in flies with varying success.Citation7 One of the first systems to solve the temporal control of transgene expression was the tetracycline (Tet)-based regulatory system. In the Tet-Off system, the tetracycline transactivator (tTA) binds the tetracycline operator (TO) constitutively, driving the expression of the transgene under the control of TO. In the presence of tetracycline, tTA is inhibited and the transgene is not expressed. The Tet-On system works in the opposite way, with tetracycline activating tTA. Both Tet-On and Tet-Off worked well in flies, although the Tet-On system required some optimization before it achieved high expression levels and more stringent response to tetracycline.Citation8 The main obstacle for the general adoption of this system is the small number of lines generated with specific expression patterns of the tTA constructs. To take advantage of the large number of Gal4 lines, the two systems were combined to induce the Tet-On activator under the control of UAS binding sites, thus responding to a spatially restricted Gal4. The disadvantage of this combined system is its inability to exploit the large number of UAS-regulated transgenes.
An alternative to the Tet-On/Off system that takes advantage of the thousands of UAS strains available is a hormone-inducible Gal4 chimeric protein. Two different systems have been generated, the most popular being a Gal4/progesterone receptor fusion known as GeneSwitch.Citation7 In this system, feeding larvae with the anti-progestin RU486 induced reporter gene expression after five hours of exposure, with maximal levels reached by 21 hours. GeneSwitch shows very low activity in early stages in the absence of the hormone, allowing the repression of toxic transgenes, although it shows some unspecific activity (leakiness) in later stages. Similar to the Tet-On system, GeneSwitch found its limitation in the small number of tissue-specific regulated driver strains. The system that takes full advantage of the tremendous popularity of the UAS/Gal4, while allowing for tight temporal control of expression, was finally developed in 2003.Citation9 TARGET (Temporal And Regional Gene Expression Targeting) consists of the introduction of a temperature-sensitive allele of the Gal4 repressor in yeasts, Gal80TS, to provide temporal control over transgene expression. This allele was introduced in flies under the control of a ubiquitously expressed tubulin promoter. When combined with the UAS/Gal4 constructs, Gal80TS demonstrated efficient, temperature dependent Gal4 regulation, with optimal repression at 19°C and derepression starting at 30°C. The dynamics of transgene activation at 32°C showed that half-maximal levels of mRNA were seen at three hours and, by six hours, mRNA levels were similar to those of the UAS/Gal4 system without Gal80TS. Thus, the TARGET system finally allows for extraordinary control of gene expression in time and space, thus providing a powerful tool to elucidate gene function.
Drosophila is also a hotbed for technical innovation in genetics and genomics. From the development of balancer chromosomes in the classic era to being one of the first organisms to be fully sequenced, this little fly has become the most versatile animal model for genetics research. Transposable elements have played a key role in the recent technological advances in flies. Initially, transposons were enigmatic elements of the genetic landscape in Drosophila and maize. Once understood, researchers exploited these mobile genetic elements to modify the genomic DNA of flies by inserting genes or other sequences of interest, including regulatory elements and artificial recombination sites.Citation10 Currently, 55% of the 13,500 genes in the Drosophila genome have been mutated with the use of transposons, a far greater percentage of stable mutant genes than in any other complex organism. The clear advantage of transposons over the more effective chemical or physical mutagens is that they leave a molecular imprint in the host gene, facilitating the task of associating a mutant phenotype with a gene. Moreover, transposon technology has experienced continuous technical advances over the last decade, resulting in increased mutagenicity. The persistent effort of a few laboratories in mobilizing transposons and identifying integration sites will produce a mutagenesis rate of >95% of the Drosophila genes in the next few years.
The constant tinkering with Drosophila transposons has produced unique resources for genetics research. One example is the ongoing effort to create a genome-wide collection of molecularly defined deletions using modified P-elements that carry FRT recombination sites.Citation11 These deletions will fill the gap between unmapped classic mutations and the wealth of genomic information available in Drosophila. Other modified transposons, carrying the UAS enhancer with a minimal promoter, have been very popular for gene discovery efforts.Citation12 Several versions of this transposon have been randomly inserted throughout the Drosophila genome, resulting in tens of thousands of invaluable strains stored in various stock centers. These insertions have been successfully used for the tissue-specific upregulation (overexpression) of nearby genes, which has identified hundreds of new genes of unknown function.
An alternative to dealing with lethal recessive mutations is the use of RNA interference (RNAi), a concept adopted from our fellow invertebrate model, the roundworm C. elegans.Citation13 Drosophila researchers understood early the advantages of RNAi for examining gene function and created a genome-wide collection of RNAi molecules for very successful cell culture assays.Citation14 More recently, this technique was expanded to in vivo studies with the creation of transgenic flies carrying RNAi constructs against every gene in the fly genome under the control of UAS. Two independent genome-wide collections are available to all interested researchers. These strains will facilitate the interrogation of the whole Drosophila genome in complex assays to rapidly advance in understanding the function of most or all of the genes in the fruit fly.
Transposon-based technology has produced remarkable advances in the manipulation of the Drosophila genome. Over the years, though, their limitations have become clear: one is the insert size they can carry (10–15 kilobases) and the second is the lack of control over insertion site, leading to undesired position effects on the transgene. To solve these problems, Drosophila researchers have imported new techniques from mouse genetics. (Yes, Drosophila researchers shamelessly steal techniques from other fields, including mouse genetics). Recombination-mediated genetic engineering or recombineering, coupled with the bacteriophage ϕC31 integrase, provides a new platform for DNA modification in flies.Citation15 This new technology provides easy and speedy DNA manipulation, finally allowing simple, straightforward homologous recombination, the missing link in the fruit fly toolbox.
Fruit Flies in the Realm of Neurodegeneration
After several decades of basic biological research, the first models of neurodegenerative diseases in Drosophila were published a decade ago.Citation16–Citation20 These papers described how expression of the mutant version of human genes associated with devastating neurological disorders reproduced relevant features of the diseases they cause in humans. These dominant neurodegenerative diseases are caused by gain-of-function mechanisms involving abnormal protein conformations, and have been easily modeled in Drosophila because these pathogenic agents maintain their neurotoxic properties in flies. For instance, the Ataxin1, Ataxin3 or Huntingtin proteins carrying polyglutamine expansions accumulate in nuclear aggregates (nuclear inclusions) in Drosophila neurons,Citation16–Citation18 whereas α-Synuclein accumulates in Lewy body-like cytoplasmic aggregates.Citation20 These pioneering works fostered the development of a new research area that has produced hundreds of research papers in the last few years. Current examples of Drosophila models of neurodegenerative disease include Alzheimer’s disease, Parkinson’s disease, tauopathies, several polyglutamine disorders (Huntington’s disease, Spinocerebellar ataxia [SCA] type 1, SCA3 [Machado-Joseph disease], Spinobulbar muscular atrophy), Amyotrophic lateral sclerosis (Lou Gehrig’s disease), non-coding expansions (SCA8, Myotonic dystrophy)Citation21 and several recessive disorders, including Fragile-X mental retardation, and dystonia, among others.
The distinctive advantage for studying human diseases in flies is to gain insight into the molecular mechanisms underlying disease pathogenesis by engaging in innovative gene discovery. In fact, fly models of several neurodegenerative disorders have realized the potential of fruit flies in the identification of genetic suppressors of neurodegeneration.Citation18,Citation22,Citation23 These efforts have confirmed the role of known pathways in protein aggregation, like the molecular chaperones and protein degradation pathways involved in quality control. These studies have also highlighted suspected pathways, like transcription factors and nuclear proteins that may be involved in the transcriptional changes described in many disorders. Finally, genetic screens have identified new pathways involved in disease pathogenesis, like RNA-binding proteins, whose role in disease is currently unknown. In a few cases, research in Drosophila has produced significant advances in dissecting the molecular mechanisms of disease. For instance, our studies in a fly model of SCA1 identified the multifunctional protein 14-3-3 as a key Ataxin1 (Atxn1) binding protein that promotes its aggregation.Citation24 The interaction of these two proteins is contingent upon Ataxin1 phosphorylation by PKB/Akt and transgenic mice carrying an amino acid substitution that prevents Atxn1 phosphorylation by Akt show no neurodegenerative changes.Citation25 The identification of these novel disease mechanisms make possible focusing on specific pharmacologic strategies to halt and/or prevent SCA1. The pharmacological approach has also been successfully tried in flies, although at a modest scale so far. Using a fly model of Huntington disease, researchers found that Histone deacetylase inhibitors suppressed cellular toxicity and arrested neuronal degeneration.Citation26 Also, compounds that inhibit mTOR and promote autophagy were neuroprotective in Drosophila models of HD and Tau neurotoxicity.Citation27,Citation28 These studies, although limited so far, demonstrate the flexibility of the Drosophila platform in studies with clear and direct implications to human health.
Modeling Prion Diseases in Flies
Following the success of these models, modeling prion diseases in flies should have been straightforward. After all, the wild type prion protein (PrP) misfolds and aggregates spontaneously in Creutzfeldt-Jakob disease (sCJD) and other prion disorders, while mutations in the Prnp gene encoding for PrP lead to familial inheritance of other prionopathies such as familial CJD, fatal familial insomnia, and Gerstmann-Sträusssler-Scheinker syndrome (GSS). However, the development of Drosophila models of prion disorders has been challenging. The first attempt to induce prion neuropathology by T. Kornberg and S. Prusiner, resulted in no degenerative effects.Citation29 Here, the authors used the Hsp70 promoter to induce expression of wild type PrP from Syrian hamster, which was activated through heat pulses. It is possible that this system could not provide sustained high levels of PrP or that the repeated heat shocks activated protective chaperones, resulting in low levels of misfolded PrP and low toxicity. A few years later, D. Harris and S. Supattapone, expressed a wild type version of mouse PrP (MoPrP) and an octarepeat mutant (PG14) associated with familial disease under the control of the UAS/Gal4 system.Citation30 Surprisingly, the flies seemed to accumulate very little mutant PrP in the brain compared with the eyes, suggesting that flies possessed a mechanism that prevented its accumulation. These two studies led to the conclusion that Drosophila was not a good model to study prion diseases. More recently, Supattapone’s group tried a different PrP mutant that leads to GSS in humans. This time they reported that expression of MoPrPP101L induced brain degeneration associated to PrP aggregation.Citation31 However, aged flies did not accumulate detergent-insoluble or PK-resistant PrP conformers, thus missing two hallmark features of pathogenic PrP. Unfortunately, a model with these characteristics would have limited practical application in the study of PrP.
Given our previous experience modeling SCA1, Huntington disease and Alzheimer’s disease in flies,Citation18,Citation24,Citation32,Citation33 we reasoned that it should be possible to create a relevant model of prion neurotoxicity in flies. We decided to develop a fly model of sporadic prion disorder, the most common type of prion disease affecting ∼80% of all patients, by expressing wild type PrP. Trying to avoid some of the pitfalls described by other investigators, we paid special attention to what we considered were the two most important issues: choosing the species of the PrP construct and inducing high expression levels. With respect to the species of PrP, we hypothesized that the accelerated disease progression seen in hamsters might be due to low conformational stability that promotes efficient conversion to pathogenic conformers. Inspired by this idea and following in Prusiner’s footsteps, we created transgenic flies expressing wild type PrP form Syrian hamster (HaPrP) under the control of UAS. Then, we selected lines expressing the highest levels of PrP as indicated by quantitative RT-PCR and western blot, and used these lines for our experiments. Interestingly, accumulation of HaPrP in the brain seemed weaker than in the eyes, supporting previous observations that there may be active mechanisms to degrade PrP in brain cells. To overcome this, we identified strong Gal4 strains expressed either in the whole brain or in specific brain areas with well-characterized morphology. Expression of wild type HaPrP with these Gal4 strains led to neuronal degeneration in the brain in just 30 days, and we observed that brain neurons undergo spongiform vacuolation by ultrastructural studies.Citation34 Next, we examined whether specific conformational changes in PrP were associated with these degenerative phenotypes. We found that wild type HaPrP undergoes progressive insolubility in the non-ionic detergent sarkosyl, a typical finding during prion conversion. Then, we showed that HaPrP experiences conformational changes comparable to those of PrPSc, as demonstrated by its resistance to a denaturing agent (guanidinium) and immunoreactivity against the 15B3 conformational antibody. However, flies did not accumulate proteinase K-resistant PrP, indicating that wild type PrP can induce spongiform degeneration in the absence of its prototypical PrPSc conformation.
After demonstrating that HaPrP misfolds and undergoes relevant conformational changes in transgenic flies, we wondered whether the most potent molecular chaperone in the cell, Hsp70, could interact and interfere with HaPrP misfolding. For this, we produced flies co-expressing HaPrP and human Hsp70 (a gift from N. Bonini) and, then, we re-examined the neurotoxicity and biochemical properties of HaPrP. In these flies, PrP neurotoxicity is reduced along with the ability of PrP to accumulate in misfolded conformations. The next question was to determine if the protective effect of Hsp70 involved a direct interaction with PrP. We confirmed this in pull-down and co-immunoprecipitation experiments.Citation34 Importantly, the Co-IP was prevented by adding ATP to the homogenate, which results in Hsp70 cycling and liberation of its substrate. These results, although interesting, led us to a paradox: how can Hsp70 interact with PrP if these two proteins occupy separate subcellular domains? Hsp70 is a cytosolic protein, while PrP is secreted and anchored to the extracellular aspect of the plasma membrane, leaving no opportunity for them to interact. Our own observations confirmed the segregation of these two proteins in microsomal preparations from young flies (day 1 post-eclosion), with all Hsp70 going to the cytosolic fraction and all PrP going with the membranes (). However, as flies aged and the conformational properties of PrP changed, the subcellular distribution of Hsp70 also changed, following PrP to the membranous domains (). In fact, we showed that in older flies expressing HaPrP, Hsp70 can colonize the lipid raft, a highly specialized domain of the plasma membrane. This observation is important because the lipid raft has been proposed to be the physical place where PrP conversion takes place.Citation35 Thus, with the use of several techniques, including locomotor behavior, histology and biochemistry, we have identified a new, powerful regulator of PrP misfolding in vivo.
Hsp70 Interferes with in vitro Conversion of Mammalian PrPSc
To further support the relevance of our recent findings in flies, we assessed the ability of Hsp70 to interfere with prion replication in a simplified mammalian system. For this, we used a modified cell-free conversion assay for PrPSc. In short, the PrPC present in a normal brain homogenate, the substrate, converts into the protease-resistant PrPSc conformer after exposure to small amounts of inoculum containing infectious brain extract. We wondered if adding exogenous recombinant Hsp70 to these experiments would alter the rate of PrP conversion. This in vitro assay is more efficient with repeated cycles of sonication (Protein Misfolding Cyclic Amplification), which increases the surface of misfolded particles (seeds).Citation36 However, we performed our amplification assays without sonication to avoid interfering with the proposed interaction between Hsp70 and PrPSc. Instead, we followed a recent protocol that only uses vigorous shaking and incubation at 37°C to achieve comparable results.Citation37 In control conditions (no exogenous Hsp70), this system produced substantial amplification of PK-resistant PrPSc after a 72-hour incubation (, lanes 1 and 2). However, when the conversion reaction took place with the addition of recombinant Hsp70, the efficiency of PrPSc conversion was significantly decreased (, lane 4). Moreover, since ATP is required to stimulate Hsp70 cycling, repeated addition of fresh ATP to the conversion reaction carrying exogenous Hsp70 inhibited prion amplification even further (, lane 3). The effect of ATP highlights the specificity of the chaperone activity of Hsp70 on PrPSc conversion, and eliminates the simple alternative of interference by the addition of an exogenous protein. Taken together, these results not only support the neuroprotective role of Hsp70 observed in flies, but also uncover a potential anti-conversion effect on mammalian prion replication.
A General Role for Hsp70 in Prion Misfolding?
So, are these results a peculiarity of the Drosophila and in vitro systems or are we describing a relevant phenomenon in prion biology? A number of observations support a central role for molecular chaperones and Hsp70 in particular in prion pathology. Hsp70 was found to be highly upregulated in the cerebellum of CJD patients.Citation38 Similarly, several targets of HSF1 (Heat shock factor 1), including Hsp70, were upregulated in mice inoculated with prions.Citation39 Furthermore, S. Lindquist has recently reported that mice lacking HSF1 activity (HSF1 knock out) die faster than wild type mice after inoculation with RML prions.Citation40 Although disease onset and PrP conversion were similar, this result provides functional basis for the role of chaperones in PrP pathogenicity in a mammalian model. Therefore, Hsp70 may play a crucial role in the pathogenesis of prion diseases and represents a new target for treating these devastating disorders.
It is also interesting to note that a cytosolic fragment of PrP (cytPrP) directly interacts with Hsp70 and prevents apoptosis in cultured neurons.Citation41 In this context, misfolded cytPrP is very toxic through its interaction with Bcl-2, although elevated levels of Hsp70 prevent this interaction. Unfortunately, it is unclear whether cytPrP has a physiological relevance. A key question in PrP biology is, thus, how can Hsp70 interact with the normal membrane-tethered PrP. A clue to this unlikely interaction comes from recent studies showing that, under stress conditions, Hsp70 can move across membranous structures and into organelles.Citation42 Indeed, Hsp70 can be released into the extra-cellular space via exosomes and can also pull proteins across membranes.Citation43 Furthermore, Hsp70 has been detected in lipid rafts in normal cells, a plasma membrane microdomain critical for PrP biology, while stress conditions exacerbate this distribution of Hsp70.Citation44 These new mechanisms of Hsp70 secretion upon specific stresses explain its direct interaction with PrP and the protective activity that we describe (see ).
Although yeast prions share conformational properties with mammalian prions, they are not completely comparable biological phenomena. For instance, yeast prions like PSI(+), PIN+ and URE3 are cytosolic proteins, whereas PrP is secreted and membrane-bound. Interestingly, many studies support a direct role of molecular chaperones in yeast prion propagation.Citation45 The barrel-shaped Hsp104 hexamers promote prion propagation by shearing the large aggregates and increasing seed surface.Citation46 However, the role of proteins of the Hsp70 family is more complex. Inducible Hsp70 collaborates with Hsp104 to promote propagation of PSI(+) prion, but it impairs URE3 prions. However, constitutive Hsp70 displays an antagonistic activity through direct interaction with PSI(+) prions. Although the function of Hsp70 in yeast prion propagation is complex and varies depending on prion type and Hsp70 isoform, a strong and direct interaction between these proteins is emerging. Combined with recent results in mammalian prions, we believe that Hsp70 plays a physiological role in PrP misfolding and transmission. It is possible that this interaction is mediated by Hsp70 co-chaperones, such as Hsp40 and Hsp90. Hsp40 directly binds substrates and presents them to the catalytic site of Hsp70. Thus, chaperone complexes that contain Hsp70 could bind PrP and directly modulate PrP conformation, stability and/or degradation in concert with the UPC (). It would be interesting to test if other families of chaperones, including the chaperonins (Hsp60) and the small chaperones (Hsp20’s), are also involved in the regulation of PrP misfolding.
Limitations of the Fly System
Although the goal of the authors is to highlight the potential contributions of fruit flies to prion pathobiology, we also want to acknowledge the limitations of this model system. An obvious limitation for modeling human diseases in flies is the reproduction of physiological disease conditions. The introduction of human genes requires the use of misexpression techniques in which the transgene may not replicate expression levels typical of the disease condition. This should be a minor problem because, while knock-in models are desirable, multiple insertions and overexpression are common features of the transgenic mice used in biomedical research. Other caveats include the evident anatomical differences, particularly in the brain, that restrict our ability to mimic the region- or neuron-specific pathology. However, the evolutionary conservation of the molecular pathways associated with learning and memory, locomotor coordination, aging, sleep, addiction and sensory processing in Drosophila and mammals support the relevance of this invertebrate model. A specific limitation of the prion models published so far is that flies do not accumulate PK-resistant PrPSc, which at present precludes transmissibility studies. On the other hand, the lack of a PrP homologue in Drosophila makes it an ideal ‘clean’, prion-free system to investigate relevant aspects of PrP misfolding and neurotoxicity. Another potential criticism for using flies in transmissibility studies is the ‘flight risk’ of material contaminated with mammalian PrPSc. The use of appropriate facilities (Arthropod Containing Laboratory level 2+) should prevent runaway (better still, flyaway) incidents. More importantly, the UAS/Gal4 regulatory system provides a key control to easily prevent PrP expression in the fly collection until a limited and carefully monitored experiment is initiated.
What the Future May Hold
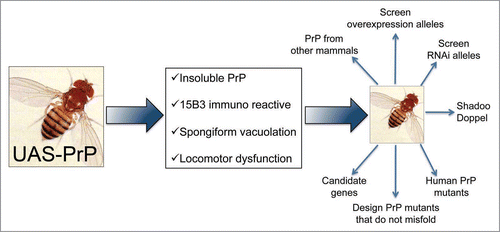
The vast genetic resources available for Drosophila research, including thousands of existing mutant strains and potent and flexible technical resources, should be a strong reason for welcoming flies into the prion community. Since flies are cheap to produce and maintain, a fly laboratory can easily escalate its operation to undertake genome-wide studies requiring the handling of several thousand (2 to 15 thousand) strains. The low cost and short life cycle of the flies also encourage risk taking, which often results in surprising discoveries (see Hsp70) and in the development of new research directions. Some predictable developments in the next few years may include testing candidate genes and unbiased screens for modifier genes ().
Candidate approach.
Drosophila strains expressing PrP can address specific questions in prion biology. For instance, a variety of PrP-binding partners have been identified over the years,Citation47 including the Laminin receptor (signal transduction), Synapsin 1b (synaptic vesicle trafficking), Calcineurin B (neuronal Ser/Thr phosphatase), Bcl-2 (anti-apoptotic function), NRAGE (apoptosis activator), N-CAM (cell adhesion) and Caveolin-1 (rafts-associated protein), to name a few. However, the biological significance of these interactions remains obscure due to the un-natural conformers used for assays and/or the kind of assay performed (yeast two-hybrid, cross-linking or co-immunoprecipitation). As D. Harris, stated, the use of a tractable genetic system such as Drosophila can play a key role in validating the physiologically relevant interactions.Citation47 Here, the flexibility of the fly system represents a unique opportunity for in vivo validation of PrP interactors as well as for the elucidation of cellular pathways and compartments in which these interactions take place ().
Unbiased screens….
Since we have established robust PrP-mediated phenotypes in flies (e.g., progressive PrP misfolding, neurodegeneration and locomotor dysfunction), it would be possible to isolate second site mutations that suppress or enhance the phenotype of choice (). This unbiased “modifier screen” would be a powerful approach to discover new molecular pathways associated with prion protein misfolding and/or pathogenesis. The advantage of such approach is that it would modify relevant aspects of PrP biology in vivo. In addition, genome-wide screens are possible thanks to the resources produced by other researchers. Thus, Drosophila represents an inexpensive and fast approach to systematically define the pathways relevant for PrP pathobiology. Finally, different schemes can be devised to take advantage of molecularly defined deletions, loss-of-function alleles (RNAi) as well as overexpression alleles, to ensure that no relevant genes are missed. These are unique resources among animal models that can lead to a deeper understanding of PrP biology.
…and beyond.
Flies could be valuable in the comparative study of different PrP mutants linked to inherited prion disorders. At least 30 single point mutations in Prnp have been associated with human diseases.Citation48 Somehow, these mutations affect the stability, processing, trafficking and/or cellular interactions of normal PrP, thus promoting PrP misfolding into pathogenic conformers and subsequent disease. However, it remains largely unknown how these different mutants lead to cell degeneration and death. The comparative analysis of these PrP mutants in the prion-free cellular environment of flies may represent a controlled experimental paradigm to assess the molecular and cellular consequences of various PrP mutations.
Transgenic flies expressing PrP could also contribute to comprehend the molecular evolution of mammalian prion proteins. PrP sequences from a significant number of mammalian species have been extensively studied in silico, but few clues have emerged about the relationship between PrP structure and disease.Citation49,Citation50 The comparative analysis of PrP from different mammals, either highly sensitive or completely resistant to prion transmission, in transgenic flies could help understand how subtle changes in the amino acid sequence of PrP determines its ability to convert and cause disease.
Methods
PrP conversion assay.
10% Brain homogenates from healthy (PrPC substrate) and infected (PrPSc inoculum) hamsters were used as described previously with minor modifications.Citation37 Briefly, 5 μl of PrPSc were added to 50 μl of PrPC in 50 μl of PBS supplemented with 0.05% SDS and 0.05% Triton X-100, 6 mM MgCl2, 1 mM EDTA, and Complete protease inhibitors. Where indicated, 1 μM of human recombinant Hsp70 or an equivalent volume of PBS 1X was added to the reaction. 6 mM of fresh ATP was also supplied every 12 hours in one reaction. One half of the conversion reaction was frozen (F) to assess efficiency of conversion. Samples were then incubated at 37°C for 72 hours with continuous, vigorous shaking. Then, PK digestion (20 µg/ml) was performed for 1 hour at 37°C and samples were analyzed by western blot, immunoblotted with 3F4 anti-PrP antibody and detected by ECL.
Conclusion
We have recently showed that transgenic flies expressing HaPrP reproduce key aspects of sporadic prion disease. Thus, we are in position to examine the genome of Drosophila for genes capable of modifying PrP misfolding and neurotoxicity. This approach can contribute to address relevant questions in PrP pathobiology, such as: Which are the molecular mechanisms triggering the spontaneous conformational changes of PrP? What are the ancillary proteins (if any) assisting in this process? Which are the molecular and cellular features associated with PrP conversion? Which are the pathogenic cascades leading to neurodegeneration? Drosophila research has the potential to produce a compendium of genetic tools to complement research in established rodent, cellular and in vitro models of PrP conversion and disease. Therefore, the future looks bright for Drosophila as an instrument to genetically dissect fundamental, unknown aspects of PrP-associated pathology.
Figures and Tables
Figure 1 Hsp70 colonizes membranous domains in response to PrP misfolding. (A) In normal conditions, PrP is synthesized in the ER, modified in the trans-Golgi network (TGN) and secreted through exosomes into the plasma membrane (PM), where it remains attached by a GPI anchor. On the other hand, Hsp70 remains in the cytosol where it contributes to nascent protein folding and protein quality control. (B) Under disease conditions, PrP accumulates in misfolded aggregates (red molecules) in the secretory pathway and the membrane. Hsp70 can detect misfolded PrP and translocates into membranous compartments to interact with PrP. Hsp70 can also be secreted to interact with PrP in lipid rafts.
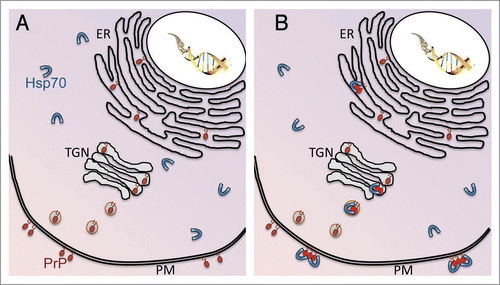
Figure 2 Hsp70 interferes with PrPSc replication in vitro. Brain homogenates from healthy hamsters were inoculated with scrapie-infected brain extracts, and incubated for 72 hours to promote conversion to PrPSc. After incubation, the reactions were digested with PK to visualize PrPSc, except for lane 5 that shows total PrP, and resolved by western blot using the 3F4 antibody. F, Equivalent aliquot of conversion reaction frozen at time zero (no amplification), with (lane 1) and without (lane 5) PK digestion. Incubation and vigorous shaking produced clear amplification of the PK-resistant isoform (lane 2). However, exogenous Hsp70 partially inhibited PrPC conversion (lane 4). This effect is ATP-dependent since addition of ATP further inhibited PrPSc accumulation (lane 3). The result shown is a representative example of four independent experiments and the ratio shown below is normalized against the amplified sample in the absence of Hsp70 (lane 2).

Figure 3 Model for Spontaneous PrP Misfolding and Hsp70 Aactivity. Flies expressing wild type PrP from hamster accumulate a harmless conformer with the biochemical properties of PrPC. Over time, PrP misfolds and converts into a neurotoxic conformation that is insoluble, resistant to denaturing agents, and contains PrPSc epitopes, but is also PK-sensitive (PrP*). This isoform is possibly an intermediary in the metabolism of infectious PrPSc in typical TSE, but it may also exist as an independent pathway. Hsp70 prevents or delays the accumulation of misfolded PrP conformers by either re-folding or tagging them for degradation by components of the Ubiquitin-Proteasome Complex (UPC).
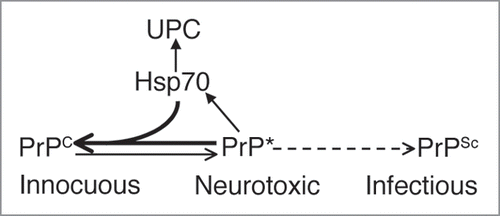
Figure 4 Possible uses for the PrP-expressing flies. Scheme for potential applications of fruit flies expressing mammalian PrP. Flies expressing HaPrP display features relevant in prion disorders, including progressive insolubility, immunoreactivity to the conformational antibody 15B3 that recognizes PrPSc conformers, spongiform vacuolation by day 30 and rapid locomotor dysfunction. These assays can be used as the basis of genetic screens to identify novel genes relevant for PrP misfolding and neurotoxicity, and to undertake complex comparative studies that may result too expensive and time consuming in transgenic mice.
Acknowledgements
We would like to thank the support of Joaquin Castilla and Claudio Soto for materials and rich conversations. This work was supported by the John Sealy Memorial Endowment Fund (CON 15431) to D.E.R.-L. and the NIH grant DP2 OD002721-01 to P.F.-F. S.C.-T. was supported by the Kempner postdoctoral fellowships.
References
- Adams MD, Celniker SE, Holt RA, Evans CA, Gocayne JD, Amanatides PG, et al. The genome sequence of Drosophila melanogaster. Science 2000; 287:16 - 20
- Venter JC, Adams MD, Myers EW, Li PW, Mural RJ, Sutton GG, et al. The sequence of the human genome. Science 2001; 291:1304 - 1351
- Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, et al. Initial sequencing and analysis of the human genome. Nature 2001; 409:860 - 921
- Matthews KA, Kaufman TC, Gelbart WM. Research resources for Drosophila: the expanding universe. Nat Rev Genet 2005; 6:179 - 193
- Brand AH, Perrimon N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development 1993; 118:401 - 415
- Rubin GM, Spradling AC. Genetic transformation of Drosophila with transposable element vectors. Science 1982; 218:348 - 353
- McGuire SE, Roman G, Davis RL. Gene expression systems in Drosophila: a synthesis of time and space. Trends Genet 2004; 20:384 - 391
- Stebbins MJ, Urlinger S, Byrne G, Bello B, Hillen W, Yin JC. Tetracycline-inducible systems for Drosophila. Proc Natl Acad Sci USA 2001; 98:10775 - 10780
- McGuire SE, Le PT, Osborn AJ, Matsumoto K, Davis RL. Spatiotemporal rescue of memory dysfunction in Drosophila. Science 2003; 302:1765 - 1768
- Venken KJ, Bellen HJ. Emerging technologies for gene manipulation in Drosophila melanogaster. Nat Rev Genet 2005; 6:167 - 178
- Ryder E, Ashburner M, Bautista-Llacer R, Drummond J, Webster J, Johnson G, et al. The DrosDel deletion collection: a Drosophila genomewide chromosomal deficiency resource. Genetics 2007; 177:615 - 629
- Bellen HJ, Levis RW, Liao G, He Y, Carlson JW, Tsang G, et al. The BDGP gene disruption project: single transposon insertions associated with 40% of Drosophila genes. Genetics 2004; 167:761 - 781
- Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, Mello CC. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 1998; 391:806 - 811
- Perrimon N, Mathey-Prevot B. Applications of high-throughput RNA interference screens to problems in cell and developmental biology. Genetics 2007; 175:7 - 16
- Venken KJ, Bellen HJ. Transgenesis upgrades for Drosophila melanogaster. Development 2007; 134:3571 - 3584
- Warrick JM, Paulson HL, Gray-Board GL, Bui QT, Fischbeck KH, Pittman RN, et al. Expanded polyglutamine protein forms nuclear inclusions and causes neural degeneration in Drosophila. Cell 1998; 93:939 - 949
- Jackson GR, Salecker I, Dong X, Yao X, Arnheim N, Faber PW, et al. Polyglutamine-expanded human huntingtin transgenes induce degeneration of Drosophila photoreceptor neurons. Neuron 1998; 21:633 - 642
- Fernandez-Funez P, Nino-Rosales ML, de Gouyon B, She WC, Luchak JM, Martinez P, et al. Identification of genes that modify ataxin-1-induced neurodegeneration. Nature 2000; 408:101 - 106
- Kazemi-Esfarjani P, Benzer S. Genetic suppression of polyglutamine toxicity in Drosophila. Science 2000; 287:1837 - 1840
- Feany MB, Bender WW. A Drosophila model of Parkinson’s disease. Nature 2000; 404:394 - 398
- Lu B, Vogel H. Drosophila models of neurodegenerative diseases. Annu Rev Pathol 2009; 4:315 - 342
- Bilen J, Bonini NM. Genome-wide screen for modifiers of ataxin-3 neurodegeneration in Drosophila. PLoS Genet 2007; 3:1950 - 1964
- Cao W, Song HJ, Gangi T, Kelkar A, Antani I, Garza D, et al. Identification of novel genes that modify phenotypes induced by Alzheimer’s beta-amyloid overexpression in Drosophila. Genetics 2008; 178:1457 - 1471
- Chen HK, Fernandez-Funez P, Acevedo SF, Lam YC, Kaytor MD, Fernandez MH, et al. Interaction of Akt-phosphorylated ataxin-1 with 14-3-3 mediates neurodegeneration in spinocerebellar ataxia type 1. Cell 2003; 113:457 - 468
- Emamian ES, Kaytor MD, Duvick LA, Zu T, Tousey SK, Zoghbi HY, et al. Serine 776 of ataxin-1 is critical for polyglutamine-induced disease in SCA1 transgenic mice. Neuron 2003; 38:375 - 387
- Steffan JS, Agrawal N, Pallos J, Rockabrand E, Trotman LC, Slepko N, et al. SUMO modification of Huntingtin and Huntington’s disease pathology. Science 2004; 304:100 - 104
- Berger Z, Ravikumar B, Menzies FM, Oroz LG, Underwood BR, Pangalos MN, et al. Rapamycin alleviates toxicity of different aggregate-prone proteins. Hum Mol Genet 2006; 15:433 - 442
- Ravikumar B, Vacher C, Berger Z, Davies JE, Luo S, Oroz LG, et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet 2004; 36:585 - 595
- Raeber AJ, Muramoto T, Kornberg TB, Prusiner SB. Expression and targeting of Syrian hamster prion protein induced by heat shock in transgenic Drosophila melanogaster. Mech Dev 1995; 51:317 - 327
- Deleault NR, Dolph PJ, Feany MB, Cook ME, Nishina K, Harris DA, et al. Post-transcriptional suppression of pathogenic prion protein expression in Drosophila neurons. J Neurochem 2003; 85:1614 - 1623
- Gavin BA, Dolph MJ, Deleault NR, Geoghegan JC, Khurana V, Feany MB, et al. Accelerated accumulation of misfolded prion protein and spongiform degeneration in a Drosophila model of Gerstmann-Straussler-Scheinker syndrome. J Neurosci 2006; 26:12408 - 12414
- Branco J, Al-Ramahi I, Ukani L, Perez AM, Fernandez-Funez P, Rincon-Limas D, et al. Comparative analysis of genetic modifiers in Drosophila points to common and distinct mechanisms of pathogenesis among polyglutamine diseases. Hum Mol Genet 2008; 17:376 - 390
- Fernandez-Funez P, Salinas J, Nino-Rosales L, Rincon-Limas DE. Hanin I, Windisch M, Poewe W, Fisher A. Novel Suppressors of Abeta Neurotoxicity. New Trends in Alzheimer and Parkinson Disorders: ADPD 2007 2007; Salzburg, Austria Medimont 37 - 41
- Fernandez-Funez P, Casas-Tinto S, Zhang Y, Gomez-Velazquez M, Morales-Garza MA, Cepeda-Nieto AC, et al. In vivo generation of neurotoxic prion protein: role for hsp70 in accumulation of misfolded isoforms. PLoS Genet 2009; 5:1000507
- Baron GS, Wehrly K, Dorward DW, Chesebro B, Caughey B. Conversion of raft associated prion protein to the protease-resistant state requires insertion of PrP-res (PrP(Sc)) into contiguous membranes. EMBO J 2002; 21:1031 - 1040
- Castilla J, Saa P, Hetz C, Soto C. In vitro generation of infectious scrapie prions. Cell 2005; 121:195 - 206
- Atarashi R, Moore RA, Sim VL, Hughson AG, Dorward DW, Onwubiko HA, et al. Ultrasensitive detection of scrapie prion protein using seeded conversion of recombinant prion protein. Nat Methods 2007; 4:645 - 650
- Kovacs GG, Kurucz I, Budka H, Adori C, Muller F, Acs P, et al. Prominent stress response of Purkinje cells in Creutzfeldt-Jakob disease. Neurobiol Dis 2001; 8:881 - 889
- Kenward N, Hope J, Landon M, Mayer RJ. Expression of polyubiquitin and heat-shock protein 70 genes increases in the later stages of disease progression in scrapie-infected mouse brjn]n. J Neurochem 1994; 62:1870 - 1877
- Steele AD, Hutter G, Jackson WS, Heppner FL, Borkowski AW, King OD, et al. Heat shock factor 1 regulates lifespan as distinct from disease onset in prion disease. Proc Natl Acad Sci USA 2008; 105:13626 - 13631
- Rambold AS, Miesbauer M, Rapaport D, Bartke T, Bjn]er M, Winklhofer KF, et al. Association of Bcl-2 with Misfolded Prion Protein Is Linked to the Toxic Potential of Cytosolic PrP. Mol Biol Cell 2006; 17:3356 - 3368
- Muchowski PJ, Wacker JL. Modulation of neurodegeneration by molecular chaperones. Nat Rev Neurosci 2005; 6:11 - 22
- Goloubinoff P, Rios Pde L. The mechanism of Hsp70 chaperones: (entropic) pulling the models together. Trends Biochem Sci 2007; 32:372 - 380
- Broquet AH, Thomas G, Masliah J, Trugnan G, Bachelet M. Expression of the molecular chaperone Hsp70 in detergent-resistant microdomjn]ns correlates with its membrane delivery and release. J Biol Chem 2003; 278:21601 - 21606
- Rikhvanov EG, Romanova NV, Chernoff YO. Chaperone effects on prion and nonprion aggregates. Prion 2007; 1:217 - 222
- Romanova NV, Chernoff YO. Hsp104 and prion propagation. Protein Pept Lett 2009; 16:598 - 605
- Westergard L, Christensen HM, Harris DA. The cellular prion protein (PrP(C)): its physiological function and role in disease. Biochim Biophys Acta 2007; 1772:629 - 644
- van der Kamp MW, Daggett V. The consequences of pathogenic mutations to the human prion protein. Protein Eng Des Sel 2009; 22:461 - 468
- van Rheede T, Smolenaars MM, Madsen O, de Jong WW. Molecular evolution of the mammalian prion protein. Mol Biol Evol 2003; 20:111 - 121
- Rongyan Z, Xianglong L, Lanhui L, Xiangyun L, Fujun F. Evolution and differentiation of the prion protein gene (PRNP) among species. J Hered 2008; 99:647 - 652