Abstract
The E200K mutation of the human prion protein (PrP) is known to cause familial Creutzfeldt–Jakob disease. In order to elucidate the effects of the mutation on the local structural stability of PrP, we performed ab initio fragment molecular orbital calculations for the wild-type human PrP and the E200K variant modeled under neutral and mild acidic conditions. The calculations revealed that this substitution markedly altered the intramolecular interactions in the PrP, suggesting that the local structural instabilities induced by the E200K mutation might cause initial denaturation of the PrP and its subsequent conversion to a pathogenic form. This work presents a new approach for quantitatively elucidating structural instabilities in proteins that cause misfolding diseases.
The key event leading to the development of Creutzfeldt-Jakob disease (CJD) is the conversion of the prion protein (PrP) from its cellular isoform to an abnormal isoform PrPSc.Citation1 However, the mechanism underlying the conversion of PrP to PrPSc and the detailed characteristics of PrPSc remain obscure. Familial CJD (fCJD) is inherited as an autosomal dominant trait that has been linked to several point mutations in the PrP. The E200K mutation, which is associated with fCJD, is of particular interest because it can be transmitted to mammals,Citation2,Citation3 and its clinical and neuropathogenic phenotypes are indistinguishable from those of sporadic CJD. The E200K variant may mimic the PrPSc structure; therefore, it is necessary to analyze this variant for elucidating the mechanism underlying the initial conversion of PrP to PrPSc.
Nuclear magnetic resonance (NMR) analysis revealed that the C-terminal globular structure of the E200K variant resembles that of the wild-type PrP (). However, it has been established that there are minor differences in the structural conformations of amino acid residues in the wild-type PrP and those in the E200K variant, and that these differences are responsible for certain differences in the electrostatic surface potentials.Citation4,Citation5 Nevertheless, the influence of these differences on the properties of the PrP remains to be resolved. It has been argued that the identification of such conformational changes may require a novel approach. Recently, it was reported that an atypical bovine spongiform encephalopathy (BSE) case harbored a mutation that was similar to the human E200K substitution, and this raised concerns whether such a mutation might cause sporadic BSE.Citation6,Citation7 This case also emphasized the importance of analyzing the E200K variant.
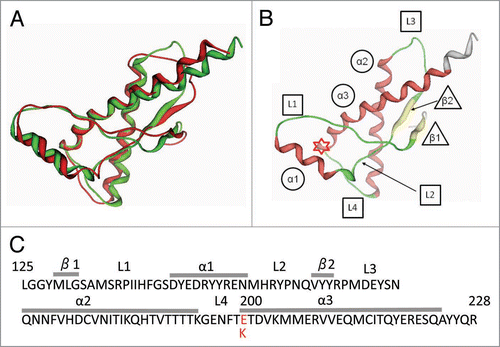
Figure 1 (A) Superposition of the 3-dimensional structures of the wild-type human PrP (green, PDB code 1QM3) and E200K variant (red, code 1OF7). (B) The secondary structure elements of human (α, α-helix; β, β-strand; L, loop). The asterisk indicates the residue at position 200 in PrP. (C) Amino acid sequence of human PrP125–228 showing secondary structure element information (α, α-helix; β, β-strand; L, loop). The asterisk indicates the residue at position 200.
Intramolecular interactions between the amino acid residues in a protein are highly dependent on the structural configurations of residue pairs, and these interactions may play a key role in the determination of the intrinsic conformations of not only the residues but also the local or global protein structures, such as secondary, tertiary or quaternary structures.Citation8,Citation9 The ab initio fragment molecular orbital (FMO) method is a promising all-electron quantum chemical procedure for estimating the molecular interactions within biomolecules.Citation10,Citation11 This method has been used to quantify the inter- and intramolecular interactions in various proteins and nucleic acids—interactions that contribute to the binding affinities and structural stabilities of these molecules.Citation12–Citation15 Very recently, this method has been successfully used to reveal the intermolecular interactions between PrP and an anti-prion drug.Citation15
In this study, we performed the FMO calculations on the globular domain of wild-type human PrP and the E200K variant in order to elucidate the differences in their intramolecular interactions and thus their local structural stabilities. This is the first study on the application of FMO calculations for investigating the intramolecular interactions in the PrP; the method represents a new approach for the structural analysis of proteins.
Results
Alternations of intra-molecular interactions at the residue 200 upon E200K mutation.
The mutation from glutamic acid to lysine markedly changed the attractive and/or repulsive interactions between adjacent amino acid residues. The selected top 10 interfragment interaction energies (IFIEs) are shown in . The negative values of IFIEs imply an attractive interaction (stable) and positive values imply a repulsive (unstable) interaction. E200, which has a negatively charged carboxylate acid side chain group, has strong attractive interaction with positively charged residues—lysine and arginine, and further, histidine in mild acidic pH—and has strong repulsive interaction with negatively charged residues—glutamic acid and aspartic acid. In contrast, K200 has attractive interaction with the negatively charged residues—glutamic acid and aspartic acid—but has repulsive interaction with the positively charged residues—lysine and arginine, and further, histidine in mild acidic pH. However, the total IFIE value between residue 200 and other residues ranged from −55 to −97 kcal/mol, and no significant difference was observed with respect to the E200K mutation. Thus, the total IFIEs of residue 200 could not reveal the difference in the structural stability between wild-type PrP and the E200K variant.
The stabilities of the secondary structure elements in PrPs: ΔEInt.
We calculated the internal interaction energies (ΔEInt) of each secondary structure element in the wild-type and variant PrPs at neutral pH in order to assess their structural stabilities (). Because ΔEInt is obtained by summing the IFIEs between the residues in the secondary structure element, in which negative IFIE values imply an attractive interaction (stable conformation) and positive values imply a repulsive (unstable) interaction, it represents the net structural stabilities of the elements. Thus, a negative ΔEInt value indicates structural stability, whereas a positive value indicates instability. The ΔEInt values were slightly lower in the variant PrP, implying that each element tends to become unstable as a consequence of the mutation. However, all the ΔEInt values in the wild-type and variant PrPs were negative, indicating that each element retained its attractive properties and was structurally stable. Although the ΔEInt for the α3 helix was relatively higher in the E200K variant (+118 kcal/mol), due to an inverse electrostatic interaction between glutamine and lysine, there was no significant difference in the structural stability of this element in the wild-type PrP and E200K variant under either neutral () or mild acidic (data not shown) pH conditions.
The stabilities of the secondary structure element pairs in PrPs: ΔEpair.
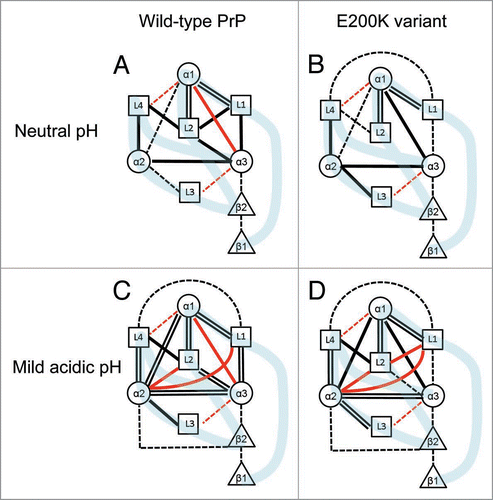
The intramolecular interaction energies (ΔEPair) of the secondary structure element pairs in the wild-type PrP and E200K variant under neutral and mild acidic conditions are shown in , and the pair interactions are illustrated in . The ΔEPair values signify the structural stability of the element pairs: a negative value indicates an attractive force between the elements and denotes a structurally stable conformation, whereas a positive value indicates repulsion and instability. It should be noted that the total ΔEPair value in the wild-type PrP was similar to that in the mutant at both neutral and acidic pH, indicating that the total structural stability of the wild-type PrP was similar to that of the mutant PrP. However, at the level of individual element pairs, certain differences in the ΔEPair values of the wild-type and E200K variant were observed. These interaction differences can be attributed to both the replacement of Glu with Lys at position 200 and the structural differences in backbone coordinates and the orientations of side chains between the wild-type PrP and the E200K variant. The strong repulsive interaction between α1 and α3 in the wild-type PrP changed to a very strong attractive interaction in the E200K variant. Under both neutral and mild acidic conditions, the wild-type and mutant PrPs exhibited very strong attractive interactions at the following sub-regions: L1-α1-L2 and L3-α2-L4. It can be suggested that these skeletal components play an important role in the maintenance of the stability of the whole PrP. Furthermore, the interactions between L1 and L2 and L2 and α3 became weak in the E200K variant (). There were very strong repulsive interactions between the L2-α2 and L1-α2 pairs in both the wild-type and variant PrPs under mild acidic conditions and very strong repulsive interaction between L2 and L1 under acidic conditions in the E200K variant ().
Figure 2 Diagrammatic representation of the intermolecular interactions in the wild-type PrP (A and C) and the E200K variant (B and D) under neutral (A and B) and mild acidic (C and D) conditions. Attractive molecular interaction energies (negative ΔE values) are shown as black lines. =, very strong attraction (|ΔE| ≥ 100 kcal/mol); —, strong attraction (100 > |ΔE| ≥ 50 kcal/mol); —, moderate attraction (50 > |ΔE| ≥ 20 kcal/mol). Repulsive interaction energies (positive δE values) are shown as red lines. —, strong repulsion (+100 > ΔE ≥ +50 kcal/mol) and —, moderate repulsion (+50 > δE ≥ +20 kcal/mol). In the diagram, weak or negligible attractions or repulsions with ΔE values less than 20 kcal/mol have not been indicated using lines. The light blue bands illustrate the connections of the backbone structures of the PrPs.
Discussion
In proteins, there exist diverse intramolecular interactions that are highly dependent on the structural conformations of not only the residues but also the secondary or tertiary structures. Such interactions are strongly associated with the structural stability of proteins.Citation8,Citation9 Thus, elucidating the intramolecular interactions in proteins would be necessary to understand the structural stability of proteins. Although the FMO calculations at the MP2 level can provide quantitative information on the molecular interactions between the residues in a protein,Citation12–Citation15 such an analysis based on individual interactions involves enormous computational complexity; for example, PrP129–224, which contains ∼100 residues, could have more than 5,000 molecular interaction energies for residue pairs. Conversion of the intramolecular interactions between residues into those for secondary structure elements or pairs dramatically reduces the computational complexity and eases the interpretation of the calculation results.
In the human PrP, the substitution of glutamine (which strongly attracts positively charged residues) at position 200 with lysine (which is functionally unlike glutamine) might cause a drastic change in the intramolecular interactions, especially for the residue at position 200 (), leading to the structural rearrangement of the PrP. However, with the exception of minor differences, the secondary and tertiary structures of the wild-type PrP and E200K variant are very similar ().Citation5 Although the FMO calculations indicated that the structural stabilities of the secondary structure elements of the wild-type PrP and E200K variant were similar (), there were considerable differences between the wild-type and variant PrP with respect to the intramolecular interactions between the pairs of the secondary elements (). Notably, the pair interactions between α1 and α3, L1 and L2, L1 and α3, and L2 and α3 in the E200K variant were different than those in the wild-type PrP ( and ). These results indicate that the E200K mutation markedly alters the local structural stability of PrP.
It has been suggested that PrPSc accumulates in the endosomes of scrapie-infected cells,Citation19 which are characterized by mild acidic conditions (pH 4.0–6.0); furthermore, it has been suggested that acidic pH might trigger the conformational transition of PrP to PrPSc.Citation20,Citation21 The calculations revealed prominent alterations in the intramolecular interactions between certain element pairs under mild acidic conditions. We focused on the element pairs with repulsive interactions (unstable conformations) specifically observed in the E200K variant because unstable pairs might be inherently related to protein denaturation. Our results suggest that the structural instabilities in the local regions around L1–L2, which are specifically observed in the E200K variant, might trigger PrP denaturation, thereby initiating its conversion to PrPSc. Therefore, by stabilizing these sub-regions, it might be possible to prevent the formation of PrPSc.
It has been proposed that the anti-prion drug, 2-pyrrolidin-1-yl-N-[4-(4-[2-pyrrolidin-1-ylacetylamino]-benzyl)-phenyl]-acetamide (GN8), is bridged to four amino acid residues of PrP [N159 and Q160 (L2), K194 (α2), and E196 (L4)], and it inhibits the conversion of PrP to PrPSc.Citation15,Citation22 Our calculations showed that the L2-α2 region with weak interactions in the neutral pH was considerably destabilized under mild acidic pH condition, although the structural stability of the L2–L4 region with strong attractive interaction in the neutral pH model did not alter considerably in the mild acidic pH ( and ). This result may suggest that the binding of GN8, especially to L2-α2 region in the endosomes, was effective in structurally stabilizing PrP, partially supporting the proposed mechanism of GN8.Citation15,Citation22
The structural stability of the entire PrP can be observed by protein denaturation measurements.Citation23 The observed thermodynamic stability of the wild-type human PrP is similar to that of the E200K variant;Citation23 this is consistent with the calculation results of the total ΔEPair values (). It was believed that the structural stability of the E200K variant was irrelevant to the conversion of PrP to PrPSc,Citation23 and that the changes in the electrostatic surface properties of PrP caused by the E200K mutation, which alters the binding characteristics of PrP, is a dominant cause of fCJD.Citation5 On the contrary, our theoretical calculations show that the intramolecular interactions in the local regions of the E200K variant were remarkably different from those in the wild-type PrP ( and ), providing a new insight into the structural instability of the E200K variant. We suggest that E200K mutation-induced alteration in the local structural stability may also play a role in the etiology of fCJD. Furthermore, electron microscopic studies have shown that PrPSc has a β-helical motif (residues 89–175), and the α2 and α3 conformation of cellular PrP is retained in PrPSc.Citation24 It can be suggested that the denaturation of a specific PrP region triggers the conversion of PrP to PrPSc. This emphasizes that the local structural instability in PrP and not global stability is important for elucidating the PrPSc conversion mechanism. Although partial unfolding in unspecified region(s) of PrP has already been monitored,Citation25 present technique could not be used to analyze the local denaturation status of PrP. It should be noted that, presently, there are no experimental data for verifying the obtained FMO calculations.
The present study demonstrates that FMO calculation is a powerful tool for quantitatively elucidating the local structural stability of proteins and has the potential to provide new insights into protein misfolding diseases. To the best of our knowledge, this is the first attempt to evaluate the intramolecular interactions in the PrP using FMO calculations.
Material and Methods
Preparation of PrP models for FMO calculations.
Initial atomic coordinates for the wild-type human PrP and E200K variant were obtained from the Protein Data Bank (PDB): codes 1QM3 and 1FO7, respectively. The wild-type PrP and E200K variant contain five major secondary structure elements: three α-helices (α1: 144–154, α2: 172–194 and α3: 200–224) and two β-strands (β1: 129–131 and β2: 161–163), as described previously.Citation5 The loop regions (L1: 132–143, L2: 155–160, L3: 164–171 and L4: 195–199) were defined as those located between the major elements, as indicated in and . The C-terminal oxygen atom was included in the wild-type models owing to the lack of relevant coordinate information. The following assumptions were made for the neutral pH model: the lysines, arginines and N terminus were considered to be positively charged; the glutamic acids, aspartic acids and C terminus were considered to be negatively charged; and histidines were considered to be neutrally protonated at the Nτ atom. All the other amino acid residues were considered to be neutral. In the mild acidic pH model, as described previously,Citation5 the protonation states were the same as in the neutral pH model, with the exception of histidines that had imidazolium side chains and thus were positively charged. The proteins were modeled in explicit water by arranging water molecules in a sphere at a distance of 10 Å from the surface of the protonated protein. After the models were optimized using an AMBER99 force field with constraints of a fixed force field for the whole protein, the water molecules were restricted to within 4.5 Å from the surface (typically, ∼250 molecules); subsequently, the protein models in water were optimized under constraints of fixed force fields for heavy atoms, with the exception of the C-terminal carboxyl group in the wild-type PrP. The modeling procedures were performed by using the molecular operating environment (MOE).Citation16
FMO calculations.
All-electron quantum chemical calculations using the ab initio FMO method were carried out using the commercial versionCitation17 of ABINIT-MP11 at the MP2/6-31G level.Citation18 The IFIE, ΔEIJ, between fragments I and J was evaluated as ΔEIJ = EIJ − EI − EJ − Tr(ΔPIJVIJ), where EIJ is the dimer electronic energy for a fragment comprising the fragments I and J; EI and EJ are monomer electronic energies for fragments I and J, respectively; and ΔPIJ and VIJ are the difference density matrix and environmental electrostatic potentialCitation11 for the IJ dimer, respectively. During the calculation, the protein structure was divided into fragments per residue unit, except for the covalently bridged disulphide residues, Cys179 and Cys214, which were treated as a single S-S bond fragment. All the interaction energies were averaged for the top three conformers in the bundle of 20 conformers deposited in the PDB. The N-terminal region 125–128 for both the wild-type and variant proteins and the C-terminal region 225–228 for the wild-type PrP and region 225–231 for the E200K variant were excluded from the molecular interaction evaluations because of their relatively high structural flexibility.
Molecular interaction analysis of the secondary structure elements in PrP.
The internal interaction energy ΔEPInt for the secondary structure element P was evaluated on the basis of the sum of all IFIEs (ΔEIJ) between the non-covalently bound residues, except for the S-S bond fragment:
The pair interaction energy between the elementsΔEPQPair between the elements P and Q was evaluated on the basis of the sum of three energy components:
where ΔEnon is the sum of the IFIEs (ΔEIJ) between the non-covalently bound residues I and J that belong to the elements P and Q, respectively. However, this equation excludes the S-S bond fragment; the molecular interaction energy contributions of this bond are taken into account in ΔESS as follows:
For the pair interaction, in which one of the element pairs P and Q is either α2 or α3, an interaction energy contribution ΔESS for the S-S bond fragment was added to ΔEPair and simply evaluated as half the interaction energy obtained from the sum of the IFIEs between the S-S bond fragment and each residue in the secondary structure element, except for α2 and α3. For the pair interaction between the covalently bridged α2 and α3, the S-S bond fragment contribution was excluded. In the case of covalently adjoining secondary structure element pairs, molecular interaction energy ΔEcov between the N-terminal and C-terminal residues in the adjoining elements was added to ΔEPair. This was simply evaluated from the IFIE between further cleaved side chain fragments or between a side chain group and a conventional fragment of corresponding pairs, in which the residues, except for glycine and threonine, were further divided into two fragments—main and side chain portions—by cutting the bond-detached atomsCitation10,Citation11 at the Cβ atom of serine, asparagine, aspartic acid, histidine, tyrosine and valine; at Cγ for glutamine and glutamic acid; and at Cδ for arginine and lysine. For valine, the interaction energy was evaluated as the sum of the IFIEs for two fragmented methyl groups.
Figures and Tables
Table 1 Selected interfragment interaction energies (IFIEs) between Glu200 in the wild-type PrP or Lys200 in the E200K variant and each amino acid residue, calculation performed at the FMO-MP2/6-31G level
Table 2 The internal interaction energies (ΔEInt) of the wild-type PrP and E200K variant modeled at neutral pH
Table 3 The pair interaction energies (ΔEPair) of the wild-type PrP and E200K variant modeled under neutral and mild acidic conditions
Acknowledgements
We are very grateful to Dr. T. Nakano, National Institute of Health Science, for his kind support. We also thank Drs. O. Hino and M. Kobayashi, AdvanceSoft Corporation, for their helpful advice. The numerical calculations were supported by the Ministry of Agriculture, Forestry and Fisheries Research Network (MAFFIN). This study was supported by a Grant-in-Aid from the BSE and other Prion Disease Control Projects of the Ministry of Agriculture, Forestry and Fisheries, Japan.
References
- Prusiner SB. Prions. Proc Natl Acad Sci USA 1998; 95:13363 - 13383
- Chapman J, Brown P, Rabey JM, Goldfarb LG, Inzelberg R, Gibbs CJ Jr, et al. Transmission of spongiform encephalopathy from a familial Creutzfeldt-Jakob disease patient of Jewish Libyan origin carrying the PRNP codon 200 mutation. Neurology 1992; 42:1249 - 12450
- Tateishi J. Transmission of human prion diseases to rodents. Seminars in Virology 1996; 7:175 - 180
- Govaerts C, Wille H, Prusiner SB, Cohen FE. Prusiner SB. Structural studies of prion proteins Prion Biology and Diseases 2004; 2nd Ed New York Cold Spring Harbor Laboratory Press 243 - 282
- Zhang Y, Swietnicki W, Zagorski MG, Surewicz WK, Sönnichsen FD. Solution structure of the E200K variant of human prion protein. Implications for the mechanism of pathogenesis in familial prion diseases. J Biol Chem 2000; 275:33650 - 33654
- Richt JA, Hall SM. BSE case associated with prion protein gene mutation. PLoS Pathog 2008; 4:1000156
- Ferguson-Smith MA, Richt JA. Rare BSE mutation raises concerns over risks to public health. Nature 2009; 457:1079
- Nishio M, Umezawa Y, Hirota M, Takeuchi Y. The CH/π interaction: significance in molecular recognition. Tetrahedron 1995; 51:8665 - 8701
- Stites WE. Protein-Protein Interactions: interface structure, binding thermodynamics and mutational analysis. Chem Rev 1997; 97:1233 - 1250
- Kitaura K, Ikeo E, Asada T, Nakano T, Uebayashi M. Fragment molecular orbital method: An approximate computational method for large molecules. Chem Phys Lett 1999; 313:701 - 706
- Nakano T, Kaminuma T, Sato T, Akiyama Y, Uebayasi M, Kitaura K. Fragment molecular orbital method: Application to polypeptides. Chem Phys Lett 2000; 318:614 - 618
- Fukuzawa K, Kitaura K, Uebayasi M, Nakata K, Kaminuma T, Nakano T. Ab initio quantum mechanical study of the binding energies of human estrogen receptor α with its ligands: An application of fragment molecular orbital method. J Comput Chem 2005; 26:1 - 10
- Fukuzawa K, Komeiji Y, Mochizuki Y, Kato A, Nakano T, Tanaka S. Intra- and intermolecular interactions between cyclic-AMP receptor protein and DNA: Ab initio fragment molecular orbital study. J Comput Chem 2006; 27:948 - 960
- Ito M, Fukuzawa K, Mochizuki Y, Nakano T, Tanaka S. Ab initio fragment molecular orbital study of molecular interactions between liganded retinoid X receptor and its coactivator; Part II: Influence of mutations in transcriptional activation function 2 activating domain core on the molecular interactions. J Phys Chem A 2008; 112:1986 - 1998
- Ishikawa T, Ishikura T, Kuwata K. Theoretical study of the prion protein based on the fragment molecular orbital method. J Comput Chem 2009; 30:2594 - 2601
- MOE version 2008 2008; Montreal, Canada Chemical Computing Group Inc.
- Advance/BioStation version 2.0 2008; Tokyo, Japan AdvanceSoft Corporation
- Mochizuki Y, Nakano T, Koikegami S, Tanimori S, Abe Y, Nagashima U, et al. A parallelized integral-direct second-order Møller-Plesset perturbation theory method with a fragment molecular orbital scheme. Theor Chem Acc 2004; 112:442 - 452
- Borchelt DR, Taraboulos A, Prusiner SB. Evidence for synthesis of scrapie prion proteins in the endocytic pathway. J Biol Chem 1992; 267:16188 - 16199
- Hornemann S, Glockshuber R. A scrapie-like unfolding intermediate of the prion protein domain PrP(121–231) induced by acidic pH. Proc Natl Acad Sci USA 1998; 95:6010 - 6014
- Kelly JW. The environmental dependency of protein folding best explains prion and amyloid diseases. Proc Natl Acad Sci USA 1998; 95:930 - 932
- Kuwata K, Nishida N, Matsumoto T, Kamatari YO, Hosokawa-Muto J, Kodama K, et al. Hot spots in prion protein for pathogenic conversion. Proc Natl Acad Sci USA 2007; 104:11921 - 11926
- Swietnicki W, Peterson RB, Gambetti P, Surewicz WK. Familial mutation and the thermodynamic stability of the recombinant human prion protein. J Biol Chem 1998; 273:31048 - 31052
- Govaerts C, Wille H, Prusiner SB, Cohen FE. Evidence for assembly of prions with left-handed β-helices into trimers. Proc Natl Acad Sci USA 2004; 101:8342 - 8347
- Morillas M, Vanik DL, Surewicz WK. On the mechanism of α-helix to β-sheet transition in the recombinant prion protein. Biochemistry 2001; 40:6982 - 6987