Abstract
It has been estimated that cerebrospinal fluid (CSF) contains approximately 80 proteins that significantly increase or decrease in response to various clinical conditions. Here we have evaluated the CSF protein PrPC (cellular prion protein) for possible increases or decreases following spinal cord injury. The physiological function of PrPC is not yet completely understood; however, recent findings suggest that PrPC may have neuroprotective properties. Our results show that CSF PrPC is decreased in spinal cord injured patients 12 hours following injury and is absent at 7 days. Given that normal PrPC has been proposed to be neuroprotective we speculate that the decrease in CSF PrPC levels may influence neuronal cell survival following spinal cord injury.
Introduction
The cellular prion protein, PrPC, is an evolutionarily conserved protein whose biological function remains elusive (reviewed in refs. Citation1 and Citation2). Following synthesis in the rough endoplasmic reticulum, PrPC is constitutively transported to the cell surface where it remains attached to the plasma membrane by a C-terminal glycosylphosphatidyl (GPI) anchor;Citation3 this localization facilitates its association with dynamic lipid raft membrane domains.Citation4 PrPC is subject to N-linked glycosylation and non-, mono- and di-glycosylated versions of PrPC are simultaneously found in the cell.Citation5 In addition to N-linked glycosylation sites, PrPC contains a single intramolecular C-terminal disulfide bond. As new PrPC is synthesized, the cell must also make triage/quality control decisions, either refolding or destroying misfolded PrPC in the proteosome or lysosome; however, the precise mechanism(s) that control steady state PrPC levels are not yet known. PrPC is highly expressed thoughout the central nervous system,Citation6 but is also found in many non-neuronal tissues such as cardiac muscle, lymphoid tissue and testes.Citation7 PrP null mice are resistant (see below) to infectious prionsCitation8 but are otherwise healthy with only subtle phenotypic abnormalities.Citation1
Prion disease such as variant Creutzfeld Jakob disease and Kuru are fatal neurodegenerative disorders associated with the conversion of the cellular prion protein (PrPC) into an alternatively folded disease specific isoform (PrPSc).Citation9 Currently, no effective treatment for prion disease is known. Either sporadic protein misfolding or discrete genetic mutations can result in the initial conversion of normal cellular PrPC, a protein of predominantly α helical structure, into a protein of predominantly β-sheet structure that is insoluble in detergents and partially resistant to protease K, PrPSc. PrPSc is a self propagating infectious protein conformation that progressively converts endogenous cellular PrPC into the abnormal PrPSc conformation.Citation9 Hence, unlike other neurodegenerative disorders (e.g., Huntington’s disease, Alzheimer disease), ingested PrPSc is believed to spread from gut to brain so that prion diseases are transmissible within species and sometimes between species (reviewed in ref. Citation10). PrPSc replication is posttranslational, however, at present there is conflicting evidence regarding the exact cellular compartment(s) in which PrPSc replicates. Expression of the normal cellular form of prion, PrPC, is essential for susceptibility to prion disease as well as replication of infectivity since PrP knockout mice are disease resistant.Citation8 Scrapie in sheep, chronic wasting disease (CWD) in elk and mule deer, bovine spongiform encephalopathy (BSE) in cattle and variant Creutzfeldt-Jacob disease (vCJD) in humans represent prion diseases that are caused by oral exposure to transmissible spongiform encephalopathy (TSE) agents. Despite concentrated efforts prompted by the threat of potential prion diseases to human health, the normal physiological function of PrPC has remained enigmatic. Moreover, the PrPSc triggered cascade of pathogenic events underlying prion-mediated neurotoxicity remains to be characterized.
Progressive misfolding of endogenous PrPC into disease associated PrPSc, excessive accumulation of prion aggregates and amyloid-like plaques, disruption of normal cellular function, neuronal loss and histopathological spongiform change are the signposts of prion disease development. Molecular chaperones have been prominent among the cellular factors speculated to influence prion disease progression. From a cell biological perspective, the initial conversion of normal cellular PrPC to the aberrant PrPSc conformation, the subsequent propagation of PrPC to PrPSc, the formation of oligomeric species/prion fibrils and the inability to eliminate toxic PrPSc can be attributed to a failure of the cellular protein quality control (i.e., the cellular chaperone machinery). Accordingly, if the collective function of cellular chaperones in health individuals is sufficient to prevent accumulation of misfolded prions, it follows that when protein quality control is compromised prion disease would progress.
Chaperones are classified into families on the basis of their molecular weight: Hsp110, Hsp70, Hsp60, Hsp40 and Hsp25. Several chaperone families have been implicated in PrPC protein homeostasis including: Hsp110,Citation11 Hsp60,Citation11,Citation12 αBcrystallin,Citation13 Rdj2,Citation14 Hsp40/Hsp70,Citation15 BiP/grp94calnexin/calreticulinCitation16 and Hsp90 (inhibitors).Citation17 In addition, given that extracellular protein deposits are associated with many neurodegenerative diseases the notion of an extracellular counter to intracellular molecular chaperones has become a recent focus of research activity (reviewed in refs. Citation18–Citation20). Although chaperones have been documented in CSF, the primary source of extracellular chaperones and the physiological significance or extracellular chaperones remain to be established. Furthermore, knockout of the transcriptional regulator of the heat shock response, HSF1 in mice result in a shorter life span than control mice after inoculation with PrPSc. This implicates stress induced chaperones in the survival of prion infected mice.Citation21 Proteins like PrPC that transit through the secretory/endocytotic pathways encounter distinct chaperones in different cellular compartments (e.g., ER resident chaperones). It follows that in a given compartment, an excess of misfolded protein or a decrease in chaperones would exacerbate protein misfolding and amyloid formation. Although chaperones are generally thought to have neuroprotective functions in maintaining protein conformation and clearing cells of misfolded proteins, it is possible that specific chaperones may override the natural energy barrier between PrPC and PrPSc to participate in prion disease.
The aim of this study was to evaluate human cerebrospinal fluid (CSF) for changes in the levels of PrPC after spinal cord injury. The CSF compartment is specialized to bathe the CNS and in addition to providing neuroprotection may be a key factor in the spread of prion disease pathogensis. Changes in the spectrum of CSF proteins reflect either changes in the biosynthesis/secretion of proteins or proteolytic degradation of proteins. Proteins may also end up in the CSF as part of cell turnover or pathological conditions (e.g., brain tumors, damage of blood-CSF barrier). Under normal conditions PrPC is released from cells into the CSF.Citation22–Citation26 We evaluated PrPC in CSF samples for two reasons: First, PrPC has been hypothesized to be neuroprotectiveCitation27–Citation38 and it follows that changes in CSF PrPC expression levels might influence neural cell survival. Secondly, CSF from some CJD patients is infectiousCitation39 and changes in the CSF levels of PrPC may well alter the rate of PrPSc replication.
Here we provide the first evidence that PrPC levels in the CSF are lower in all spinal cord injured patients compared to control patients and that the reduction was PrPC specific. We also examined CSF for components of the traditional cellular quality control machinery, which may be important for the maintenance of prion conformation and neuroprotection. Hsp70, Hsc70, Hsp60 were absent in all CSF samples including neurological controls. Our results indicate that many traditional intracellular protein quality control pathways are absent in CSF following spinal cord injury and we speculate that CSF proteins like PrPC are relatively vulnerable to misfolding resulting in the accumulation of potentially toxic species in this compartment. Surprisingly, members of the Hsp27 family, which are characterized by the presence of a homologous sequence of 80–100 residues, referred to as the α-crystallin domain, were notably increased after spinal cord injury (within 12 h of surgery) in some CSF samples. Our findings identify CSF as a unique compartment that can undergo large changes in PrPC and Hsp27s levels.
Results and Discussion
PrPC is reduced in CSF after spinal cord injury. In order to investigate the possibility that CSF levels of PrPC change, we evaluated PrPC levels in the CSF of patients after spinal cord injury. CSF contains a wealth of diagnostic/prognostic information. shows the western blot analysis of PrPC in human CSF from a control patient, and a patient at 12 h, 24 h, 2, 3, 4, 5 and 6 d after spinal cord injury. PrPC is detected by Western analysis as 28 kDa, 32 kDa and a dominant 37 kDa PrPC immunoreactive band, representing the un-, mono- and di-glycosylated PrPC species respectively that are not completely resolved in . PrPC in CSF may be from (a) either damaged cells or (b) hydrolysis of the GPI anchor. further shows PrPC levels were lower in four spinal cord injured patients compared to control patients and that the 37 kDa band is the major species in the CSF samples. Samples taken within 12 h following injury already had lower values than controls (no injury) and PrPC was often not detectable at 7 d following spinal cord injury. Reduction in CSF levels of PrPC after spinal cord injury was confirmed with two distinct antibodies (6H4 Prionics and 11C6 SPI-BIO). shows the clinical data from six controls and 13 spinal cord injured patients. We found that minocycline treatment did not alter reduction of PrPC. Tetracycline has been reported to decrease prion infectivity in animal models in other studies.Citation40 Variations among the CSF levels of PrPC were observed between patients, but in all cases PrPC was reduced following spinal cord injury. Taken together, these results confirm previous studies documenting PrPC in human CSFCitation24,Citation41,Citation42 and further show that PrPC levels in CSF can be subject to variations in response to trauma. We specifically show that spinal cord injured patients have reduced levels of PrPC in CSF.
14-3-3 is not a marker for spinal cord injury. Next we examined CSF for the presence of the cytoplasmic protein, 14-3-3 (). 14-3-3 is an abundant protein of the central nervous system and the presence of 14-3-3 proteins in CSF samples is a clinical biomarker for extensive and rapid neuronal destruction (e.g., prion disease, dementia).Citation43 This diagnostic test is currently used as the leading preliminary screen for prion diseases; however, it is used in combination with other diagnostic criteria due to the overlap between prion diseases and other conditions that elevate CSF levels of 14-3-3. None of the CSF samples evaluated, from either control or spinal cord injured patients, were found to have detectable 14-3-3 protein. Note that there are multiple 14-3-3 isoforms expressed in human brain and these experiments evaluated 14-3-3beta/zeta isoforms. Our results indicate that the presence of 14-3-3 is not a clinical biomarker for spinal cord injury. In contrast, CSF levels of IgG heavy chain (55 kDa) and light chain (26 kDa) were observed to increase following spinal cord injury. demonstrates that IgG light chain increases in CSF after spinal cord injury. We evaluated the antibodies present in the CSF patient samples for PrPC immunoreactivity. CSF antibodies were not immunoreactive with PrPC (data not shown).
A CSF expression of crystallin-domaincontaining- proteins is variable in patients following spinal cord injury. In contrast, demonstrates that members of the Hsp27 family were identified in some (but not all) CSF samples. αA/αB-crystallin immunoreactive proteins were found in several CSF samples. Also, Hsp27 immunoreactive bands were found in several CSF samples. Significant differences in Hsp25/27 expression in the brain and spinal cord is reported between mice and rats.Citation44,Citation45 In contrast to cellular prion protein, Hsp27 and αA/αB-crystalin levels in CSF did not correlate with spinal cord injury. αA/αB-crystallin has been implicated in the prevention of protein aggregation and fibril formation in a wide range of diseases. Although a direct PrPC::αB-crystallin association has been documented,Citation13 our results indicate that CSF levels of αA/αB-crystallin do not decrease like that observed for PrPC following spinal cord injury and that αA/αB-crystallin levels in the CSF are variable between patients following spinal cord injury (). Taken together these results indicate that following spinal cord injury CSF levels of PrPC decrease, while 14-3-3 levels remain unchanged and IgG levels increase. In contrast to PrPC, αA/αB-crystallin, another protein implicated in neuroprotection, does not show a consistent reduction in CSF levels following spinal cord injury.
Hsp40s, Hsc70, Hsp90 and Hsp110 are absent from CSF obtained from control and spinal cord injured patients. The relevance of molecular chaperones to diseases involving protein misfolding remains a central biological question. In addition to prion diseases a number of other protein conformation disorders are associated with amyloid deposits (e.g., Alzheimer disease, Down syndrome)Citation20 in the absence of proteosome inhibition. The traditional view is that chaperones interact with hydrophobic residues associated with partially unfolded unstructured chain segments, to guard against various folding traps such as aggregation and other nonproductive interactions that result in disease. Current evidence suggests that different chaperones follow distinct strategies to fold nacent proteins, protect, refold mature proteins and eliminate misfolded proteins. Once the native conformation is achieved, chaperones are thought to dissociate from the protein. While typically thought to be neuroprotective, it is possible that chaperones may protect proteins that contribute to diseases processes (e.g., toxic conformations or conformations that elicit an autoimmune response). To begin to address the role of protein chaperones in maintaining function we evaluated CSF of patients after spinal cord injury for Hsp40, Rdj2, Hdj2, Hsp110, Hsp90, Hsp70, Hsc70 and Hsp60. shows that these traditional cellular chaperones were not found in the CSF of spinal cord injured patients. ATP levels in CSF are lower than cellular levels,Citation20 further suggesting that the ATP dependent intracellular chaperone machinery that PrPC encounters during its transit through the secretory/endocytotic pathway does not maintain PrPC conformation in the CSF. Taken together, although several members of the traditional ATP-dependent chaperone machinery have been implicated in PrPC conformational regulation,Citation19 with the exception of the Hsp27 family (see below), they do not appear to play a role in CSF in the patients evaluated in this study.
Methods
Subject enrollment. Between 2004 and 2009 subjects presenting with spinal cord injury (SCI) with motor deficit to the Foothills Hospital spine service and presenting within 12 h of their injury were offered enrolment in a clinical trial examining minocycline and spinal cord perfusion pressure (SCPP) augmentation (ClinicalTrials.gov identifier NCT00559494). Following informed consent, all subjects received a lumbar indwelling catheter for measurement of spinal fluid pressure and for sampling of CSF. Surgical decompression and stabilization was performed within 24 h of injury. Of note, no subjects were treated with corticosteroids. All protocols were approved by the University of Calgary Conjoint Health Research Ethics Board.
Minocycline treatment. Subjects were randomized (1:1) to intravenous minocycline or placebo. The first 10 subjects were randomized to placebo (equivolume of normal saline) versus minocycline (Wyeth Pharmaceuticals) 200 mg intravenously twice daily. Subsequent subjects were randomized to placebo versus minocycline 800 mg intravenous loading dose followed by twice daily dosing tapering each dose by 100 mg to 400 mg that was then maintained for the remainder of time. Study treatment was continued for 7 days.
Perfusion pressure augmentation. An indwelling lumbar catheter inserted at the L4/5 level was transduced to measure spinal cerebrospinal fluid pressure. An indwelling arterial catheter placed in the radial artery at the wrist was similarly transduced for blood pressure. SCPP (mean arterial pressure minus cerebrospinal fluid pressure) was electronically monitored in real time. If the SCPP fell below 75 mmHg at any time during the first seven days, (except during a surgical procedure), the patient was randomly assigned to guideline directed blood pressure management (control) versus SCPP augmentation. Subjects assigned to control were administered crystalloid fluid and if necessary inotrope therapy (norepinephrine) to maintain mean arterial pressure above 65 mmHg. Subjects assigned to SCPP augmentation were administered crystalloid fluid and if necessary inotrope therapy (norepinepherine) to maintain SCPP greater than 75 mmHg. Treatment was continued until the end of day 7.
CSF collection. CSF samples (up to 10 mL each) were collected from the indwelling lumbar catheter starting at the time of enrolment before first administration of the study intervention and for the subsequent 7 d scheduled around the twice daily study drug administration (30 min infusion) as follows: ½ hour before drug infusion, ½ hour after drug administration and 6 h after drug administration essentially providing samples every 6 h. Samples were centrifuged at 2,000 rpm for 5 min to separate cellular matter and the supernatant was flash frozen and stored at −80°C in aliquots.
Neurological control samples (10 mL) were collected from six subjects by lumbar puncture under general anaesthesia prior to microdiscectomy surgery. These individuals had evidence of symptomatic degenerative disc disease refractory to non-surgical management for greater than 12 wks but no evidence of central nervous system pathology on history and physical exam.
Sample analysis. Prior to all sample preparation procedures, all samples were blinded. Forty microliters of each CSF sample was fractionated by SDS-PAGE and transferred from polyacrylamide gels to nitrocellulose (0.45 µm) in 20 mM Tris, 150 mM glycine and 12% methanol. Membranes were blocked with 4% milk solution (prepared in PBS with 0.1% Tween 20) and incubated with primary antibody for 2 h at room temperature or overnight at 4°C. The membranes were washed in blocking solution and incubated with horseradish peroxidase-coupled secondary antibody. The signal was developed using West Pico Pierce reagent (Pierce Biotechnology, Inc.,) and exposed to Kodak film.
Cytosolic and membrane fractions of rat brains were prepared as previously describedCitation49 and PC12 cells were prepared as previously described.Citation50
Antibodies. Anti-PrPC monoclonal were from Prionics (6H4) or SPI-BIO (11C6) as indicated. Anti-Hsp70 mouse monoclonal (SPA 810), anti-Hsp110 rabbit polyclonal (SPA 1101), anti- Hsp40 rabbit polyclonal (SPA 400), anti-Hsp90 rat monoclonal (SPA 835), anti-Hsp25 rabbit polyclonal (SPA 801), anti-Hsp27 mouse monoclonal (SPA 800), anti-Hsp20 rabbit polyclonal (SPA 803), anti-14-3-3 mouse monoclonal (KAM CC012E) and αA/αB-crystallin were from Assay Designs. Anti-Hsp70/Hsc70 mouse monoclonal (H5147) and Hsp60 (H4149) was from Sigma.
Summary
A main observation of this study is that based on CSF analysis of PrPC, spinal cord injured patients could clearly be distinguished from neurological controls that show no evidence of central nervous system pathology. While suggested roles for PrPC include cell signaling, differentiation and adhesion, an increasing number of studies have implicated PrPC in neuroprotection against oxidative stress, ischemia and toxic protein aggregation.Citation27–Citation36,Citation38,Citation46 Given the proposed neuroprotective role for PrPC, low levels of PrPC in CSF following spinal cord injury may well create a window in which neurons are susceptible to damage. CSF reduction in PrPC following spinal cord injury is specific ( and ). For example, expression of proteins with αA/αB crystallin domains were observed to increase, decrease or remain unchanged following spinal cord injury depending on the patient. What regulates PrPC levels in CSF? The primary determinants of the level of PrPC in the CSF are not yet established but almost certainly involve a balance between folding, trafficking, release, retrotranslocation and degradation. Future investigations will establish if CSF levels of PrPC are reduced in neurological situations other than spinal cord injury. In agreement with our study, Meyne et al.Citation47 recently showed that total prion levels are reduced in the CSF in patients with a number of neurological diseases. Cellular levels of PrPC are subject to rapid change, for example PrPC is reported to be upregulated after focal cerebral ischemia34 and disparities between the PrP mRNA and cellular PrPC protein expression have been documented.Citation48 Understanding the precise mechanisms that underlie abrupt changes in the levels of PrPC in CSF, such as the large reduction observed after spinal cord injury, should offer significant insights into the physiological role of prions.
The CSF compartment is specialized to bathe the CNS and thereby has the capacity to deliver both neuroprotective proteins (e.g., PrPC) and toxic proteins that transmit diseases (e.g., PrPSc). CSF from some CJD patients is infectious,Citation39 indicating the presence of the toxic prion template in human CSF. However, infectivity of human CSF from CJD patients is not 100%. It is tempting to speculate that changes in the levels of PrPC in CSF may support this range of sensitivity to PrPSc infection. While high levels of PrPC in CSF may be neuroprotective as outlined above, it would also support the accumulation of the pathogenic isoform. This raises the possibility that reducing PrPC in CSF would reduce sensitivity to PrPSc infection. Furthermore, Castagna et al.Citation24 have previously documented extensive PrPC fragmentation using two-dimensional electrophoresis of human CSF. Their data indicate that measurements of the full length proteins (e.g., ) underestimates total PrPC in CSF. These PrPC fragments differ in glycosylation and N terminal truncation and are proposed to be generated during the extracellular/intracellular cycling of PrPC a process also implicated in the neuronal spread of toxic prions. It is possible that extensive PrPC fragmentation would influence PrPSc’s infectivity. Thus, in the case of prion disease, changes in CSF levels of PrPC may well influence disease progression following PrPSc exposure, however there is currently no direct support for this notion. Future studies will have to explore whether high CSF levels of PrPC do in fact effect prion disease progression.
Figures and Tables
Figure 1 PrPC levels in human CS F are attenuated following spinal cord injury. (A) Western analysis of PrPC in human CS F from control (lane1) and from spinal cord injured patient #19 (remaining lanes) at 12 h, 24 h, day 2, day 3, day 4, day 5 and day 6 following spinal cord injury. 40 µl of the indicated CS F samples were separated by SDS PAGE, the Western blot was probed with anti-PrPC 6H4 (Prionics) antibody and developed with SuperSignal® West Dura (Pierce). (B) Western analysis of representative human CS F from control, 12 h within spinal cord injury and 7 d after spinal cord injury (40 µl). Four patients are shown. Blots were probed with anti-PrPC 6H4 (Prionics) antibody or anti-PrPC 11C6 antibody (SPI-BIO).
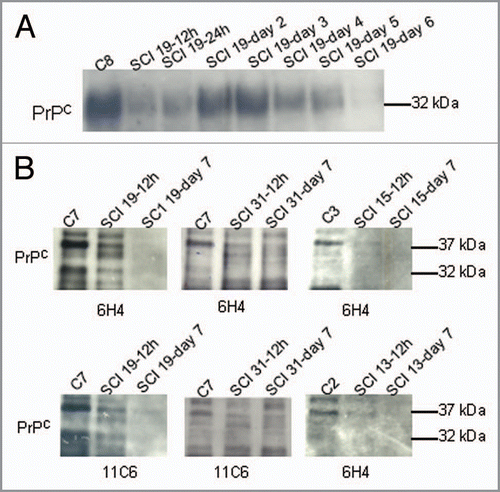
Figure 2 14-3-3 and IgG light chain levels in CS F after spinal cord injury. (A) Western analysis of 14-3-3 and IgG light chain after spinal cord injury. Three patients are shown. 14-3-3 is absent in all CS F samples. IgG light chains are present in varying amounts in all samples. 20 µg of rat brain supernatant and pellet fractions and 20 µg of PC12 cell homogenate (middle) are shown in the right lanes. (B) Western analysis of CS F (40 µl) after spinal cord injury for αA/αB crystallin and Hsp27.
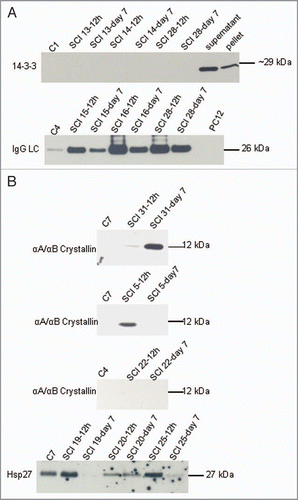
Figure 3 Molecular chaperones were not detected in CSF of control or spinal cord injured patients. Western analysis of Hsp40, Rdj-2, Hdj-2, Hsp110, Hsp90, Hsc70, Hsp70 and Hsp60 in CS F samples (40 µl) from control or spinal cord injured patients as indicated. 20 µg of rat brain supernatant and pellet fractions are shown in the right lanes.
Table 1 Clinical characteristics of the patients included in the study
Acknowledgements
We thank Tatania Noskova and Claudia Silva for technical contributions. This work was funded by the Alberta Prion Research Institute. J.E.A.B. is an Alberta Heritage Research Foundation for Medical Research Senior Scholar.
References
- Watts JC, Westaway D. The prion protein family: diversity, rivalry and dysfunction. Biochim Biophys Acta 2007; 1772:654 - 672
- Gains MJ, LeBlanc AC. Canadian Association of Neurosciences Review: prion protein and prion diseases: the good and the bad. Can J Neurol Sci 2007; 34:126 - 145
- Stahl N, Borchelt DR, Hsiao K, Prusiner SB. Scrapie prion protein contains a phosphatidylinositol glycolipid. Cell 1987; 51:229 - 240
- Naslavsky N, Stein R, Yanai A, Friedlander G, Taraboulos A. Characterization of detergent-insoluble complexes containing the cellular prion protein and its scrapie isoform. J Biol Chem 1997; 272:6324 - 6331
- Haraguchi T, Fisher S, Olofsson S, Endo T, Groth D, Tarentino A, et al. Asparagine-linked glycosylation of the scrapie and cellular prion proteins. Arch Biochem Biophys 1989; 274:1 - 13
- Sales N, Rodolfo K, Hassig R, Faucheux B, Di Giamberardino L, Moya KL. Cellular prion protein localization in rodent and primate brain. Eur J Neurosci 1998; 10:2464 - 2471
- Ford MJ, Burton LJ, Morris RJ, Hall SM. Selective expression of prion protein in peripheral tissues of the adult mouse. Neuroscience 2002; 113:177 - 192
- Büeler H, Aguzzi A, Sailer A, Greiner RA, Autenried P, Aguet M, et al. Mice devoid of PrP are resistant to scrapie. Cell 1993; 73:1339 - 1347
- Prusiner SB. Novel proteinaceous infectious particles cause scrapie. Science 1982; 216:136 - 144
- Davies GA, Bryant AR, Reynolds JD, Jirik FR, Sharkey KA. Prion diseases and the gastrointestinal tract. Can J Gastroenterol 2006; 20:18 - 24
- DebBurman SK, Raymond GJ, Caughey B, Lindquist S. Chaperone-supervised conversion of prion protein to its protease-resistant form. Proc Natl Acad Sci USA 1997; 94:13938 - 13943
- Edenhofer F, Rieger R, Famulok M, Wendler W, Weiss S, Winnacker EL. Prion protein PrPc interacts with molecular chaperones of the Hsp60 family. J Virol 1996; 70:4724 - 4728
- Sun G, Guo M, Shen A, Mei F, Peng X, Gong R, et al. Bovine PrPc directly interacts with alphaB-crystalline. FEBS Lett 2005; 579:5419 - 5424
- Beck KE, Kay JG, Braun JE. Rdj2, a J protein family member, interacts with cellular prion PrP(C). Biochem Biophys Res Commun 2006; 346:866 - 871
- Rambold AS, Miesbauer M, Rapaport D, Bartke T, Baier M, Winklhofer KF, et al. Association of Bcl-2 with misfolded prion protein is linked to the toxic potential of cytosolic PrP. Mol Biol Cell 2006; 17:3356 - 3368
- Capellari S, Zaidi SI, Urig CB, Perry G, Smith MA, Petersen RB. Prion protein glycosylation is sensitive to redox change. J Biol Chem 1999; 274:34846 - 34850
- Ochel HJ, Gademann G, Trepel J, Neckers L. Modulation of prion protein structural integrity by geldanamycin. Glycobiology 2003; 13:655 - 660
- Calderwood SK, Mambula SS, Gray PJ Jr, Theriault JR. Extracellular heat shock proteins in cell signaling. FEBS Lett 2007; 581:3689 - 3894
- Tytell M, Robinson MC, Milligan CE. Release of Heat Shock Proteins and their Effects When in the Extracellular Space in the Nervous System 2008; Springer
- Wilson MR, Yerbury JJ, Poon S. Extracellular Chaperones and Amyloids. Asea AA, Brown IR, editors. Springer. Heat Shock Proteins and the Brain: Implications for Neurodegenerative Diseases and Neuroprotection. Asea AA and Calderwood SK. Ref Type: Serial (Book, Monograph) 2008; 3:283 - 315
- Steele AD, Hutter G, Jackson WS, Heppner FL, Borkowski AW, King OD, et al. Heat shock factor 1 regulates lifespan as distinct from disease onset in prion disease. Proc Natl Acad Sci USA 2008;
- Picard-Hagen N, Gayrard V, Viguie C, Moudjou M, Imbs C, Toutain PL. Prion protein in the cerebrospinal fluid of healthy and naturally scrapie-affected sheep. J Gen Virol 2006; 87:3723 - 3727
- Vella LJ, Greenwood DL, Cappai R, Scheerlinck JP, Hill AF. Enrichment of prion protein in exosomes derived from ovine cerebral spinal fluid. Vet Immunol Immunopathol 2008; 124:385 - 393
- Castagna A, Campostrini N, Farinazzo A, Zanusso G, Monaco S, Righetti PG. Comparative two-dimensional mapping of prion protein isoforms in human cerebrospinal fluid and central nervous system. Electrophoresis 2002; 23:339 - 346
- Stahl N, Borchelt DR, Prusiner SB. Differential release of cellular and scrapie prion proteins from cellular membranes by phosphatidylinositol-specific phospholipase C. Biochemistry 1990; 29:5405 - 5412
- Fevrier B, Vilette D, Archer F, Loew D, Faigle W, Vidal M, et al. Cells release prions in association with exosomes. Proc Natl Acad Sci USA 2004; 101:9683 - 9688
- Kuwahara C, Takeuchi AM, Nishimura T, Haraguchi K, Kubosaki A, Matsumoto Y, et al. Prions prevent neuronal cell-line death. Nature 1999; 400:225 - 226
- McLennan NF, Brennan PM, McNeill A, Davies I, Fotheringham A, Rennison KA, et al. Prion protein accumulation and neuroprotection in hypoxic brain damage. Am J Pathol 2004; 165:227 - 235
- Spudich A, Frigg R, Kilic E, Kilic U, Oesch B, Raeber A, et al. Aggravation of ischemic brain injury by prion protein deficiency: role of ERK-1/-2 and STAT-1. Neurobiol Dis 2005; 20:442 - 449
- Weise J, Doeppner TR, Müller T, Wrede A, Schulz-Schaeffer W, Zerr I, et al. Overexpression of cellular prion protein alters postischemic Erk1/2 phosphorylation but not Akt phosphorylation and protects against focal cerebral ischemia. Restor Neurol Neurosci 2008; 26:57 - 64
- Shyu WC, Lin SZ, Chiang MF, Ding DC, Li KW, Chen SF, et al. Overexpression of PrPc by adenovirusmediated gene targeting reduces ischemic injury in a stroke rat model. J Neurosci 2005; 25:8967 - 8977
- Mitteregger G, Vosko M, Krebs B, Xiang W, Kohlmannsperger V, Nölting S, et al. The role of the octarepeat region in neuroprotective function of the cellular prion protein. Brain Pathol 2007; 17:174 - 183
- Lee KJ, Panzera A, Rogawski D, Greene LE, Eisenberg E. Cellular prion protein (PrPc) protects neuronal cells from the effect of huntingtin aggregation. J Cell Sci 2007; 120:2663 - 2671
- Weise J, Crome O, Sandau R, Schulz-Schaeffer W, Bahr M, Zerr I. Upregulation of cellular prion protein (PrPc) after focal cerebral ischemia and influence of lesion severity. Neurosci Lett 2004; 372:146 - 150
- Bounhar Y, Zhang Y, Goodyer CG, LeBlanc A. Prion protein protects human neurons against Bax-mediated apoptosis. J Biol Chem 2001; 276:39145 - 39149
- Walz R, Amaral OB, Rockenbach IC, Roesler R, Izquierdo I, Cavalheiro EA, et al. Increased sensitivity to seizures in mice lacking cellular prion protein. Epilepsia 1999; 40:1679 - 1682
- Shmerling D, Hegyi I, Fischer M, Blättler T, Brandner S, Götz J, et al. Expression of amino-terminally truncated PrP in the mouse leading to ataxia and specific cerebellar lesions. Cell 1998; 93:203 - 214
- Nishida N, Tremblay P, Sugimoto T, Shigematsu K, Shirabe S, Petromilli C, et al. A mouse prion protein transgene rescues mice deficient for the prion protein gene from purkinje cell degeneration and demyelination. Lab Invest 1999; 79:689 - 697
- Brown P, Gibbs CJ Jr, Rodgers-Johnson P, Asher DM, Sulima MP, Bacote A, et al. Human spongiform encephalopathy: the National Institutes of Health series of 300 cases of experimentally transmitted disease. Ann Neurol 1994; 35:513 - 529
- Forloni G, Iussich S, Awan T, Colombo L, Angeretti N, Girola L, et al. Tetracyclines affect prion infectivity. Proc Natl Acad Sci USA 2002; 99:10849 - 10854
- Wong BS, Green AJ, Li R, Xie Z, Pan T, Liu T, et al. Absence of protease-resistant prion protein in the cerebrospinal fluid of Creutzfeldt-Jakob disease. J Pathol 2001; 194:9 - 14
- Tagliavini F, Prelli F, Porro M, Salmona M, Bugiani O, Frangione B. A soluble form of prion protein in human cerebrospinal fluid: implications for prion-related encephalopathies. Biochem Biophys Res Commun 1992; 184:1398 - 1404
- Yutzy B, Holznagel E, Coulibaly C, Stuke A, Hahmann U, Deslys JP, et al. Time-course studies of 14-3-3 protein isoforms in cerebrospinal fluid and brain of primates after oral or intracerebral infection with bovine spongiform encephalopathy agent. J Gen Virol 2007; 88:3469 - 3478
- Armstrong CL, Krueger-Naug AM, Currie RW, Hawkes R. Constitutive expression of the 25-kDa heat shock protein Hsp25 reveals novel parasagittal bands of purkinje cells in the adult mouse cerebellar cortex. J Comp Neurol 2000; 416:383 - 397
- Armstrong CL, Krueger-Naug AM, Currie RW, Hawkes R. Constitutive expression of heat shock protein HSP25 in the central nervous system of the developing and adult mouse. J Comp Neurol 2001; 434:262 - 274
- Khosravani H, Zhang Y, Tsutsui S, Hameed S, Altier C, Hamid J, et al. Prion protein attenuates excitotoxicity by inhibiting NMDA receptors. J Cell Biol 2008; 181:551 - 565
- Meyne F, Gloeckner SF, Ciesielczyk B, Heinemann U, Krasnianski A, Meissner B, et al. Total prion protein levels in the cerebrospinal fluid are reduced in patients with various neurological disorders. J Alzheimers Dis 2009; 17:863 - 873
- Ford MJ, Burton LJ, Li H, Graham CH, Frobert Y, Grassi J, et al. A marked disparity between the expression of prion protein and its message by neurones of the CNS. Neuroscience 2002; 111:533 - 551
- Rosales-Hernandez A, Beck KE, Zhao X, Braun AP, Braun JE. RDJ2 (DNAJA2) chaperones neural G protein signaling pathways. Cell Stress Chaperones 2008; 14:71 - 82
- Gibbs SJ, Barren B, Beck KE, Proft J, Zhao X, Noskova T, et al. Hsp40 couples with the CSPalpha chaperone complex upon induction of the heat shock response. PLoS ONE 2009; 4:4595