Abstract
Prions are self-propagating proteinaceous infectious agents capable of transmitting disease in the absence of nucleic acids. The nature of the infectious agent in prion diseases has been at the center of passionate debate for the past 30 years. However, recent reports on the in vitro generation of prions have settled all doubts that the misfolded prion protein (PrPSc) is the key component in propagating infectivity. However, we still do not understand completely the mechanism of prion replication and whether or not other cellular factors besides PrPSc are required for infectivity. In this article, we discuss these recent reports under the context of the protein-only hypothesis and their implications.
Prions, Bizarre Infectious Agents, Unique Diseases
Prions are self-propagating particles of proteinaceous origin which share the ability to transmit disease with typical infectious organisms such as viruses and bacteria, but in contrast to them, prions do not have genetic material.Citation1,Citation2 Prion diseases have been found in humans and other mammals, including cattle, sheep, cervids, felines and rodents. In addition to transmission by infection, the disease can have inherited and sporadic origins. In the transmissible cases, infection of the host is preceded by a variable incubation period and followed by the appearance of clinical symptoms. Prion diseases are 100% fatal and after a long pre-symptomatic period in which the agent is slowly replicating, the clinical phase is often very rapid, progressive and severe.Citation3
Although the exact molecular nature of prions is not completely clear, it is widely accepted in the field that the prion protein (denoted PrP) in its infectious conformation (PrPSc) is the main, or perhaps only, component of the infectious agent.Citation2,Citation3 Having a misfolded protein as an infectious agent makes prions very unconventional. Even if some co-factors are proven to be required, the minimal infectious agent should be much less complex than a virus or any other form of conventional micro-organism. Given the heretical nature of prions, the “protein-only” hypothesis has remained controversial for decades.Citation4,Citation5 Recent reports demonstrating the formation of highly infectious prions completely in vitro have provided the strongest proof for the prion hypothesis and have taken the field to an entirely new position.Citation6–Citation9 In this article, we summarize these findings and critically discuss their contribution to understand the molecular nature of prions and their unique mechanism of propagation.
In Vitro Formation of Synthetic Prions
For many years the prion skeptics argued that the final proof for the prion hypothesis will be the generation of infectious material in the test tube, in the absence of living cells and hopefully with highly purified synthetic PrP.Citation5 This goal attempted by many groups failed until recently. describes some of the successful experiments reporting the production of infectious prions de novo.
An approach that was extensively explored for the purpose of generating de novo infectious material consisted on producing PrP with mutations associated with inherited prion diseases. The rationale for this approach is that the mutation should favor the formation of the infectious folding. Several mutant PrPres-like molecules have been generated, some of which were shown to acquire various biochemical properties of PrPres. However, so far, none of them have been shown to be infectious when the mutant protein produced in cells was inoculated into animals.Citation10–Citation12 Nevertheless, recent experiments from the groups of Lindquist and Aguzzi have shown that transgenic mice expressing a mutation associated to fatal familial insomnia or modifications inducing rigidization of the loop at position 166–175 of PrP, respectively, develop spontaneous disease that can be transmissible to wild type animals.Citation13,Citation14 Although these studies are certainly a step ahead on showing that infectivity is encoded in the PrP molecule, they do not address the issue of infectivity generation in vitro.
Another widely explored strategy consisted of using a variety of physicochemical procedures to induce the misfolding of recombinant protein or short PrP synthetic peptides into β sheet-rich structures exhibiting some of the biochemical and biological properties of PrPres.Citation15–Citation20 These experiments have largely failed in producing infectivity. However, in 2004, Legname and co-workers showed that a recombinant mouse PrP fragment (residues 89–230) assembled into amyloid fibrils produced disease with some prion characteristics when injected into transgenic mice highly overexpressing the same PrP sequence.Citation21 The disease was later transmitted to wild-type animals in a second passage. These findings and subsequent studies from the same group,Citation22–Citation24 including the formation of many novel synthetic prions and even some composed of protease-sensitive PrP, have provided strong evidence that PrPSc is the only element needed for infectivity. However, since the disease was originally transmitted to transgenic animals overexpressing the PrP gene and not to wild-type animalsCitation24 it cannot be ruled out that the effect seen might just be an acceleration of the disease process that was set to occur spontaneously at a later time. Indeed, it is well known that transgenic mice overexpressing PrP develop a prion-like disease spontaneously,Citation25–Citation27 including the animals used in the original Legname et al. publication. Citation24 In addition, the clinical and histopathological presentation of the disease was different from the usual disease in mice. The authors argued that this result may be due to the creation of a new strain of prions.Citation21
In a very recent study from Baskakov’s group it was reported that prion infectivity was generated in wild type hamsters after serial inoculation with full-length rPrP that had been converted into cross-β-sheet amyloid fibrils and subjected to annealing in the presence of normal brain homogenate or albumin.Citation28 However, no disease was produced in the first passage although PrPres was detected in the brain of some of the animals. Serial transmission gave rise to a disease phenotype with highly unique clinical and neuropathological features, including deposition of large amyloid plaques and a very slow progression of disease after onset of clinical signs.
In summary, the attempts to produce infectious material de novo from rPrP have only produced partial success in generating disease, because either especial transgenic mice need to be used as hosts or the disease does not appear in a first passage. These findings suggest that the infectious folding might be a very unique conformation that we are still far from mastering in the test tube.
In Vitro Generation of Prions by PMCA
In 2001, our group developed a technique to replicate prions in vitro, termed Protein Misfolding Cyclic Amplification (PMCA).Citation29 PMCA improved the efficiency of prion conversion over the pioneer work of Caughey with the cell-free conversion process,Citation30,Citation31 enabling to test the infectious properties of in vitro generated PrPSc. Considering the many failures on the attempts to produce infectious folding from rPrP, we hypothesized that this conformation must be very special and thus we attempted to mimic the pathological process of prion formation in the test tube. PMCA was shown to reproduce the auto-catalytic replication of prions in a greatly accelerated manner.Citation29 The in vitro-generated material was shown to keep all the biological, biochemical and structural characteristics of in vivo produced prions.Citation6 More importantly, we showed for the first time the production of bona-fide infectivity in wild type animals.Citation6 Recently, we have shown that different prion strains can retain their properties with high fidelity after many passages in PMCA.Citation32 Furthermore, more complex processes, such as species barrier crossing and strain adaption, were recreated in our lab using this technology.Citation33,Citation34 These findings indicate that infectivity and associated properties (strain variability, species barrier and strain adaptation) are purely dependent on the formation of PrPSc, which can be done in a cell-free system, thus ruling out the involvement of genetic material.
Using the PMCA technology, a landmark study from Supattapone’s group showed that infectious prions can be propagated with highly purified components with the sole addition of synthetic polyanions.Citation8 Strikingly, they did not only amplify a 263K strain using purified mammalian PrPC as a template, but they also observed that PrPSc molecules were randomly formed in the negative controls after a larger number of PMCA cycles.Citation8 These de novo generated prions (not seeded by preexisting PrPSc) were shown to be infectious in wild type hamsters. Clinical symptoms, neuropathological and biochemical characteristics of the disease observed largely resembled those associated to the 263K strain.Citation8 Although it is not surprising that de novo generated hamster prions look like the 263K strain, since the hamster PrP sequence has predilection for this particular form, these results raise the possibility that spontaneously generated prions may have originated by cross-contamination. However, the authors took many precautions to rule out the possibility of cross-contamination.Citation8
The possibility that prions can be spontaneously formed and amplified by PMCA is sound and feasible, considering that a large proportion of prion diseases have a sporadic origin. It is thought that sporadic prion disease are produced by a spontaneous low-frequency event of misfolding that originates the first stable molecule of PrPSc, which then grows up by the infectious mechanism. In a recent work, our group showed that de novo formation of prions can be induced under certain experimental conditions using PMCA.Citation7 Previously, we have observed on rare occasions that PMCA negative controls (healthy brain homogenates subjected to PMCA) became positive after many PMCA rounds in a random fashion. In an attempt to dig deeper into this issue, we started modifying several conditions such as buffers, addition of chaotropic agents, pH and temperature changes with the aim of facilitating this de novo process of PrPSc formation. None of these methods worked satisfactorily.Citation7 Only when the length of PMCA cycling was increased did we observe spontaneous PrPSc-like protease resistant bands. This material was infectious to wild type hamsters and generated a disease with unique clinical, neuropathological and biochemical properties.Citation7 Since the publication of this article, we have generated various novel prion strains in several species. It is astonishing to observe the large variety of prion strains that can be generated in vitro, indicating that the strain diversity is almost unlimited. However, it is important to note that some species appear to have a tendency to adopt some specific strains, e.g., the hamster PrP sequence has a good predilection for strains of the 263K or Hyper type. Therefore, the de novo formation of prions opens up a great opportunity to explore the natural tendency of a defined sequence to generate a specific prion strain. Most experimental strains have been the result of years of in vivo passages and adaptation and they carry information from heterogeneous origins. It would be interesting to see what the natural propensities of different PrP sequences are.
However, both in our studies as well as in Deleault et al.’s, the PrPC substrate came from brain origin. Atarashi et al. reported the optimization of PMCA using rPrP,Citation35 but have not shown yet whether the PrPSc produced in vitro is infectious. In a recent and exciting development, Wang et al. reported the de novo generation of bonafide infectious prions in vitro by PMCA using exclusively rPrP with the sole addition of RNA and lipids.Citation9 Again, although the material produced has the typical characteristics of prions, the disease produced indicates a novel prion strain. This study represents the so far strongest proof for the prion hypothesis.
Is PrPSc the Only Component of the Infectious Agent?
The tremendous progress in the in vitro generation of infectious prions produced in the past five years has dissipated all doubts with respect to the validity of the prion hypothesis. Today the contention that prions might be composed by a virus, bacteria or any other type of traditional micro-organism is untenable. However, it is still not possible to definitively rule out that components other than the protein are an integral part of the infectious agent. Indeed, in the studies of Deleault et al. and Wang et al. it was necessary to add non-coding RNA and possibly lipids in order to generate infectivity.Citation8,Citation9 If PrPSc is the sole component of the infectious agent, why do all these experiments require the presence of co-factors that seem to remain as stable components of the infectious particle?Citation36 This is an important question for the near future. It is possible that the requirement of RNA (or other synthetic polyanions) and lipids is needed for efficient in vitro amplification by PMCA, instead of a need of these compounds for infectivity. Indeed, in our experience, as well as in the studies published by Supattapone, a co-factor plays an important role in prion replication in vitro.Citation37,Citation38 The identification of the co-factor operating in the brain (which in vitro can be substituted by RNA, polyanions and lipids) may provide a novel and important target for prion therapy.
The prion phenomenon of transmission of biological information by “infectious proteins” in the absence of nucleic acids has also been demonstrated to operate in several yeast and fungal proteins.Citation39–Citation41 Diverse genetic, biochemical and structural evidence have been provided in support of the prion nature of various yeast proteins.Citation41,Citation42 It has been shown that bacterially produced N-terminal fragments of Sup35p, when transformed into amyloid fibrils, were able to propagate the prion phenotype to yeast cells.Citation43–Citation45 Infection of yeast with different conformers led to generation of distinct strains in vivo, indicating that differences in the conformation of the infectious protein determine prion strain variation.Citation44,Citation46 The spontaneous formation of prions has been also reproduced in yeast prions.Citation47,Citation48 However, the way spontaneous yeast prions are formed and induced experimentally differs from those of mammalian prions. The introduction of a plasmid overexpressing a particular prion protein in yeast has proven to be very efficient in generating de novo appearance of prion-associated phenotypes, which have been shown to be transmissible.Citation47,Citation49 In addition, no cofactors are needed when using recombinant proteins to reconstitute infectivity in vitro.Citation43–Citation45
Theoretical Considerations About the Mechanism of Prion Formation
Spontaneous formation of self—propagating proteinaceous particles can be explained in terms of a stochastic conformational change that renders the protein prone to, first, aggregate in an amyloidlike manner, and second, acquire seeding capabilities. The oligomerization is key to produce a stable unit and the acquisition of seeding capability is essential for infectivity transmission. In a very simplistic view, a protein with prion-like potential would possess a rather rugged energy landscape in which different prion-prone conformers may coexist at once. The fact that spontaneous population of these states remains a very rare event, based on the very low spontaneous appearance of prion-associated phenotypes in a population of individuals, can be explained by two alternative scenarios:
The thermodynamic model in which energetically non favorable states would be highly unpopulated due to the presence of much lower energy, more probable states which dominate the overall protein energy landscape. Considering the aggregate-like nature of prions, the critical misfolding event and the subsequent formation of a minimal infectious particle would require the productive clash of several monomers in the already very low-populated state, giving rise to a very infrequent phenomenon. However, considering the high stability of prions, it follows that this very low populated state would be either an intermediate state between PrPC and PrPSc (often called PrP*)Citation50 or a PrPSc-like monomer that would acquire stability upon oligomerization.
The kinetic model in which prionprone conformers might not be necessarily separated from native states by high energy differences, but by rather large activation energy barriers. These high activation energies would prevent the protein from sampling potentially prion-like alternative states.
Of course, we are currently far from determining which process controls prion formation, but both scenarios capture well various experimental observations. For instance, the very low frequency of these events in vivo can be explained from both thermodynamic and kinetic views. In the thermodynamic view, the difference lies in the fact that energetically non-favored states attain rapid equilibrium, allowing for the presence of prion-prone conformers coexisting with native states at all times, whereas in the kinetic scenario, population of these states may require days, weeks or even years depending upon the energetic gap. The probability of a productive encounter of two improbable prion-prone conformers, though it can in principle be similar in both scenarios, will be established at virtually any time in the thermodynamic view, whereas very large activation energies will preclude molecules from sampling these conformational states, unless sufficient time is provided. We also need to consider that oligomerization processes take place while misfolding occurs, making any thermodynamic or kinetic analysis much more complex in terms of quantification as well as energy landscape representations.
The thermodynamic scenario falls a little short when considering the many failed attempts to generate infectious recombinant PrP by modulating its folding in the absence of co-factors. Within the thermodynamic framework, the use of chemical and/or physical agents to reshape the PrP energy landscape should have already yielded infectious PrP if only very low populated conformers were solely required for generating prions spontaneously. Indeed, when we tried to generate de novo prions in brain homogenates, longer incubation times and many passages in PMCA reactions were effective in stimulating formation of spontaneous prions, instead of the use of chemical reagents.Citation7 We think that the high number of passages and long incubation times needed to generate spontaneous prions more likely represent a kinetic framework for PrPSc formation. Deleault et al. and Wang et al. also showed stochastic formation of prions after many rounds of PMCA using purified components.Citation8,Citation9
A kinetically-controlled process for spontaneous PrPSc formation is also in agreement with the fact that in the cell, PrPc has been estimated to have a turnover of a few hours.Citation51–Citation53 This time would be enough to keep the protein far away from sampling PrPSc-like states. However, this scenario still poses some difficulties when trying to rationalize a mechanistic framework for PrP misfolding. Timescales of the dynamics of protein conformational changes typically fall in the range of microseconds to seconds. Very slow refolding kinetics can be associated to proline isomerization as has been recently suggested,Citation54 which has also been shown to be important in amyloid formation.Citation55,Citation56 Although this process is considered to be very slow in a molecule timescale, it usually falls in the order of seconds, which makes it difficult to reconcile with the PrPSc timescale of in vitro spontaneous formation. An exquisite combination of specific proline isomeratization steps in two or more interacting PrP monomers may be underlying the process, which in addition to the nucleation process may cover up for the long time required for prion formation.
The Structure of the Infectious Folding: The Last Frontier
To date, the molecular structure of PrPSc is unknown and it represents the main big unanswered question in the field. Several models have been proposed,Citation57–Citation61 but much more work is required to resolve this important issue. PrPC is composed of two domains, a natively unfolded N-terminal domain and a globular C-terminal domain.Citation62 Although there is still debate about the structural fate of PrP C-terminal region during conversion into a prion,Citation63 it is nowadays widely accepted that part of the N-terminal domain (specifically from residues in between 89–145) acquire significant structure during misfolding.Citation64,Citation65 Of the available models, the β-helical representation seems to fit better with many different experimental constraints.Citation59 This model proposes that part of the N-terminal region folds into a β-helix forming a trimmer, as the basic structural unit. Interestingly, folding of other β-helix proteins has been shown to be an extremely slow process, even slower than proline isomerization-based folding. Two well studied cases of β-helix folding mechanism, Pertactin and P22 tailspike protein, have shown to exhibit very long folding times in vitro, probably the longest reported so far.Citation66–Citation68 The β-helix fold in these proteins is achieved in the monomer itself, as opposed to PrP which seems to form a multimeric β-helix. The lack of nucleating structural elements such as α-helices in β-helix folds is perhaps responsible for the very low folding rates. The chance of misfolding is also very high due to the high β-sheet content present on the final structure. Indeed, aggregation and formation of fibers have been reported for the nonamyloidogenic β-sheet P22 tailspike protein. Citation69 There is also compelling evidence indicating that co-translational folding of these proteins avoids aggregation in vivo, suggesting again a high propensity of this fold to promote aggregation.Citation70 Remarkably, the β-helix fold of Pertactin also exhibits partial resistance to proteolytic digestion.Citation67 The self-propagation and seeding capability of prions would then be a consequence of providing missing or low populated β-strands to normally folded PrPC as in a donor-strand complementation process.
Based on these theoretical considerations, we propose a model in which PrP misfolds into a β-helix or similar conformation that requires very long incubation times rather than partially denaturing conditions (). This implies a kinetic mechanistic framework for prion formation
Conclusion and Future Directions
The prion field has seen an explosive progress in the past five years. Thanks to the development of techniques to replicate prions in vitro with high efficiency, most of the key milestones pending to prove definitively the hypothesis that the infectious agent is purely composed by a protein have been achieved. Indeed, PrPSc has been shown to self-replicate its misfolding at expenses of highly purified PrPC in the absence of living cells;Citation8,Citation30 bonafide infectivity has been amplified millions of times by cyclic amplification of PrPSc;Citation6,Citation71 de novo generation of infectivity has been achieved both with purified mammalian and recombinant PrPC with the sole addition of RNA and lipids;Citation8,Citation9 the strain diversity, species barrier and strain adaptation processes have been mimicked in vitro by replication of prions in the test tube.Citation32–Citation34
In spite of this impressive progress, still some key questions are pending, namely: Is PrPSc the only component of the infectious material? Or does it needs the presence of non-coding accessory molecules such as polyanions and lipids? If the later is correct, what is the role of these accessory molecules in infectivity? Finally, an area where much work is still required is understanding the molecular mechanism and forces involved in prion formation and the detailed tridimensional structure of PrPSc. Therefore, although we can safely consider that the prion hypothesis has been proven, the prion field is still full of promising areas of research.
Abbreviations
| PrP | = | prion protein |
| rPrP | = | recombinant prion protein |
| PrPSc | = | misfolded PrP |
| PrPc | = | cellular PrP |
| PrPres | = | protease-resistant PrP |
| PMCA | = | protein misfolding cyclic amplification |
Figures and Tables
Figure 1 Diagram of multiple species in equilibrium with PrPC. Depending on the solution conditions, PrPC can form different types of aggregates. Amorphous aggregates arise from nonspecific protein aggregation pathways through the denatured state. Under partially denaturing conditions, PrPc can also form amyloid-type of structures which appear not to be infectious in animal models. The formation of PrPSc is depicted as an exquisite time-dependent misfolding process with the putative presence of an intermediate. The bell-shaped curves represent the apparent high energy barriers precluding PrPC from forming infectious aggregates under normal conditions. These barriers may be associated to complex processes such as β-helix folding and nucleation. The presence of co-factors such as poly-anions, lipids and yet unknown molecules can also modulate these reactions.
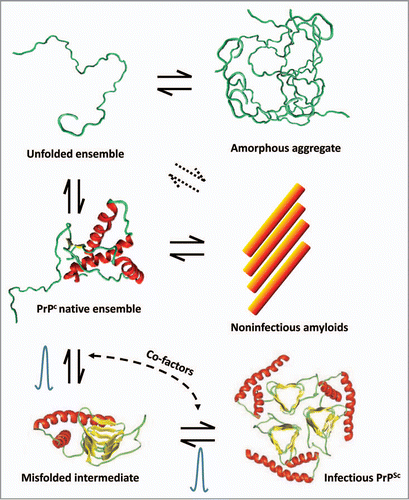
Table 1 Strategies successfully used for de novo generation of infectious prions in vitro
Acknowledgements
This work was supported in parts by NIH grants R01 NS049173 and P01 IA077774 to C.S.
References
- Prusiner SB. Novel proteinaceous infectious particles cause scrapie. Science 1982; 216:136 - 144
- Prusiner SB. Prions. Proc Natl Acad Sci USA 1998; 95:13363 - 13383
- Aguzzi A, Calella AM. Prions: protein aggregation and infectious diseases. Physiol Rev 2009; 89:1105 - 1152
- Aguzzi A, Polymenidou M. Mammalian prion biology: one century of evolving concepts. Cell 2004; 116:313 - 327
- Soto C, Castilla J. The controversial protein-only hypothesis of prion propagation. Nat Med 2004; 10:63 - 67
- Castilla J, Saá P, Hetz C, Soto C. In vitro generation of infectious scrapie prions. Cell 2005; 121:195 - 206
- Barria MA, Mukherjee A, Gonzalez-Romero D, Morales R, Soto C. De novo generation of infectious prions in vitro produces a new disease phenotype. PLoS Pathog 2009; 5:1000421
- Deleault NR, Harris BT, Rees JR, Supattapone S. Formation of native prions from minimal components in vitro. Proc Natl Acad Sci USA 2007; 104:9741 - 9746
- Wang F, Wang X, Yuan C-G, Ma J. Generating a prion with bacterially expressed recombinant prion protein. Science 2010; 327:1132 - 1135
- Lehmann S, Harris DA. Two mutant prion proteins expressed in cultured cells acquire biochemical properties reminiscent of the scrapie isoform. Proc Natl Acad Sci USA 1996; 93:5610 - 5614
- Chiesa R, Piccardo P, Ghetti B, Harris DA. Neurological illness in transgenic mice expressing a prion protein with an insertional mutation. Neuron 1998; 21:1339 - 1351
- Barron RM, Manson JC. A gene-targeted mouse model of P102L Gerstmann-Straussler-Scheinker syndrome. Clin Lab Med 2003; 23:161 - 173
- Jackson WS, Borkowski AW, Faas H, Steele AD, King OD, Watson N, et al. Spontaneous generation of prion infectivity in fatal familial insomnia knockin mice. Neuron 2009; 63:438 - 450
- Sigurdson CJ, Nilsson KP, Hornemann S, Heikenwalder M, Manco G, Schwarz P, et al. De novo generation of a transmissible spongiform encephalopathy by mouse transgenesis. Proc Natl Acad Sci USA 2009; 106:304 - 309
- Jackson GS, Hosszu LL, Power A, Hill AF, Kenney J, Saibil H, et al. Reversible conversion of monomeric human prion protein between native and fibrilogenic conformations. Science 1999; 283:1935 - 1937
- Baskakov IV, Legname G, Baldwin MA, Prusiner SB, Cohen FE. Pathway complexity of prion protein assembly into amyloid. J Biol Chem 2002; 277:21140 - 21148
- Bocharova OV, Breydo L, Parfenov AS, Salnikov VV, Baskakov IV. In vitro conversion of full-length mammalian prion protein produces amyloid form with physical properties of PrP(Sc). J Mol Biol 2005; 346:645 - 659
- Cobb NJ, Apetri AC, Surewicz WK. Prion protein amyloid formation under native-like conditions involves refolding of the C-terminal alpha-helical domain. J Biol Chem 2008; 283:34704 - 34711
- Jones EM, Surewicz WK. Fibril conformation as the basis of species- and strain-dependent seeding specificity of mammalian prion amyloids. Cell 2005; 121:63 - 72
- Lee S, Eisenberg D. Seeded conversion of recombinant prion protein to a disulfide-bonded oligomer by a reduction-oxidation process. Nat Struct Biol 2003; 10:725 - 730
- Legname G, Baskakov IV, Nguyen HO, Riesner D, Cohen FE, DeArmond SJ, et al. Synthetic mammalian prions. Science 2004; 305:673 - 676
- Legname G, Nguyen HO, Baskakov IV, Cohen FE, DeArmond SJ, Prusiner SB. Strain-specified characteristics of mouse synthetic prions. Proc Natl Acad Sci USA 2005; 102:2168 - 2173
- Colby DW, Giles K, Legname G, Wille H, Baskakov IV, DeArmond SJ, et al. Design and construction of diverse mammalian prion strains. Proc Natl Acad Sci USA 2009; 106:20417 - 20422
- Colby DW, Wain R, Baskakov IV, Legname G, Palmer CG, Nguyen HO, et al. Protease-sensitive synthetic prions. PLoS Pathog 2010; 6:1000736
- Chiesa R, Drisaldi B, Quaglio E, Migheli A, Piccardo P, Ghetti B, et al. Accumulation of protease-resistant prion protein (PrP) and apoptosis of cerebellar granule cells in transgenic mice expressing a PrP insertional mutation. Proc Natl Acad Sci USA 2000; 97:5574 - 5579
- Westaway D, DeArmond SJ, Cayetano-Canlas J, Groth D, Foster D, Yang SL, et al. Degeneration of skeletal muscle, peripheral nerves, and the central nervous system in transgenic mice overexpressing wild-type prion proteins. Cell 1994; 76:117 - 129
- Castilla J, Gutierrez-Adan A, Brun A, Pintado B, Parra B, Ramirez MA, et al. Different behavior toward bovine spongiform encephalopathy infection of bovine prion protein transgenic mice with one extra repeat octapeptide insert mutation. J Neurosci 2004; 24:2156 - 2164
- Makarava N, Kovacs GG, Bocharova O, Savtchenko R, Alexeeva I, Budka H, et al. Recombinant prion protein induces a new transmissible prion disease in wild-type animals. Acta Neuropathol 2010; 119:177 - 187
- Saborio GP, Permanne B, Soto C. Sensitive detection of pathological prion protein by cyclic amplification of protein misfolding. Nature 2001; 411:810 - 813
- Kocisko DA, Come JH, Priola SA, Chesebro B, Raymond GJ, Lansbury PT, et al. Cell-free formation of protease-resistant prion protein. Nature 1994; 370:471 - 474
- Caughey B. Formation of protease-resistant prion protein in cell-free systems. Curr Issues Mol Biol 2000; 2:95 - 101
- Castilla J, Morales R, Saa P, Barria M, Gambetti P, Soto C. Cell-free propagation of prion strains. EMBO J 2008; 27:2557 - 2566
- Castilla J, Gonzalez-Romero D, Saa P, Morales R, De CJ, Soto C. Crossing the species barrier by PrP(Sc) replication in vitro generates unique infectious prions. Cell 2008; 134:757 - 768
- Green KM, Castilla J, Seward TS, Napier DL, Jewell JE, Soto C, et al. Accelerated high fidelity prion amplification within and across prion species barriers. PLoS Pathog 2008; 4:1000139
- Atarashi R, Moore RA, Sim VL, Hughson AG, Dorward DW, Onwubiko HA, et al. Ultrasensitive detection of scrapie prion protein using seeded conversion of recombinant prion protein. Nat Methods 2007; 4:645 - 650
- Geoghegan JC, Valdes PA, Orem NR, Deleault NR, Williamson RA, Harris BT, et al. Selective incorporation of polyanionic molecules into hamster prions. J Biol Chem 2007; 282:36341 - 36353
- Deleault NR, Lucassen RW, Supattapone S. RNA molecules stimulate prion protein conversion. Nature 2003; 425:717 - 720
- Abid K, Morales R, Soto C. Cellular factors implicated in prion replication. FEBS Lett 2010; 3:2409 - 2411
- Wickner RB. [URE3] as an altered URE2 protein: evidence for a prion analog in Saccharomyces cerevisiae. Science 1994; 264:566 - 569
- Wickner RB, Taylor KL, Edskes HK, Maddelein ML, Moriyama H, Roberts BT. Prions of yeast as heritable amyloidoses. J Struct Biol 2000; 130:310 - 322
- Uptain SM, Lindquist S. Prions as protein-based genetic elements. Annu Rev Microbiol 2002; 56:703 - 741
- Wickner RB, Taylor KL, Edskes HK, Maddelein ML, Moriyama H, Roberts BT. Prions in Saccharomyces and Podospora spp.: protein-based inheritance. Microbiol Mol Biol Rev 1999; 63:844 - 861
- King CY, Diaz-Avalos R. Protein-only transmission of three yeast prion strains. Nature 2004; 428:319 - 323
- Tanaka M, Chien P, Naber N, Cooke R, Weissman JS. Conformational variations in an infectious protein determine prion strain differences. Nature 2004; 428:323 - 328
- Brachmann A, Baxa U, Wickner RB. Prion generation in vitro: amyloid of Ure2p is infectious. EMBO J 2005; 24:3082 - 3089
- Diaz-Avalos R, King CY, Wall J, Simon M, Caspar DL. Strain-specific morphologies of yeast prion amyloid fibrils. Proc Natl Acad Sci USA 2005; 102:10165 - 10170
- Masison DC, Maddelein ML, Wickner RB. The prion model for [URE3] of yeast: spontaneous generation and requirements for propagation. Proc Natl Acad Sci USA 1997; 94:12503 - 12508
- Lancaster AK, Bardill JP, True HL, Masel J. The spontaneous appearance rate of the yeast prion [PSI+] and its implications for the evolution of the evolvability properties of the [PSI+] system. Genetics 2010; 184:393 - 400
- Ross CD, McCarty BR, Hamilton M, Ben-Hur A, Ross ED. A promiscuous prion: efficient induction of [URE3] prion formation by heterologous prion domains. Genetics 2009; 183:929 - 940
- Harrison PM, Chan HS, Prusiner SB, Cohen FE. Thermodynamics of model prions and its implications for the problem of prion protein folding. J Mol Biol 1999; 286:593 - 606
- Caughey B, Race RE, Ernst D, Buchmeier MJ, Chesebro B. Prion protein biosynthesis in scrapie-infected and uninfected neuroblastoma cells. J Virol 1989; 63:175 - 181
- Borchelt DR, Scott M, Taraboulos A, Stahl N, Prusiner SB. Scrapie and cellular prion proteins differ in their kinetics of synthesis and topology in cultured cells. J Cell Biol 1990; 110:743 - 752
- Harris DA. Trafficking, turnover and membrane topology of PrP. Br Med Bull 2003; 66:71 - 85
- Baskakov IV. Switching in amyloid structure within individual fibrils: implication for strain adaptation, species barrier and strain classification. FEBS Lett 2009; 583:2618 - 2622
- Eakin CM, Berman AJ, Miranker AD. A native to amyloidogenic transition regulated by a backbone trigger. Nat Struct Mol Biol 2006; 13:202 - 208
- Smajlovic A, Berbic S, Schiene-Fischer C, Tusek-Znidaric M, Taler A, Jenko-Kokalj S, et al. Essential role of Pro 74 in stefin B amyloid-fibril formation: dual action of cyclophilin A on the process. FEBS Lett 2009; 583:1114 - 1120
- Wille H, Michelitsch MD, Guenebaut V, Supattapone S, Serban A, Cohen FE, et al. Structural studies of the scrapie prion protein by electron crystallography. Proc Natl Acad Sci USA 2002; 99:3563 - 3568
- Wille H, Bian W, McDonald M, Kendall A, Colby DW, Bloch L, et al. Natural and synthetic prion structure from X-ray fiber diffraction. Proc Natl Acad Sci USA 2009; 106:16990 - 16995
- Govaerts C, Wille H, Prusiner SB, Cohen FE. Evidence for assembly of prions with left-handed {beta}-helices into trimers. Proc Natl Acad Sci USA 2004; 101:8342 - 8347
- Cobb NJ, Sonnichsen FD, McHaourab H, Surewicz WK. Molecular architecture of human prion protein amyloid: a parallel, in-register beta-structure. Proc Natl Acad Sci USA 2007; 104:18946 - 18951
- DeMarco ML, Daggett V. From conversion to aggregation: protofibril formation of the prion protein. Proc Natl Acad Sci USA 2004; 101:2293 - 2298
- Riek R, Hornemann S, Wider G, Glockshuber R, Wuthrich K. NMR characterization of the full-length recombinant murine prion protein, mPrP(23–231). FEBS Lett 1997; 413:282 - 288
- Ji HF, Zhang HY. beta-sheet constitution of prion proteins. Trends Biochem Sci 2010; 35:129 - 134
- Shindoh R, Kim CL, Song CH, Hasebe R, Horiuchi M. The region approximately between amino acids 81 and 137 of proteinase K-resistant PrPSc is critical for the infectivity of the Chandler prion strain. J Virol 2009; 83:3852 - 3860
- Peretz D, Williamson RA, Matsunaga Y, Serban H, Pinilla C, Bastidas RB, et al. A conformational transition at the N terminus of the prion protein features in formation of the scrapie isoform. J Mol Biol 1997; 273:614 - 622
- Junker M, Clark PL. Slow formation of aggregation-resistant beta-sheet folding intermediates. Proteins 2010; 78:812 - 824
- Junker M, Schuster CC, McDonnell AV, Sorg KA, Finn MC, Berger B, et al. Pertactin beta-helix folding mechanism suggests common themes for the secretion and folding of autotransporter proteins. Proc Natl Acad Sci USA 2006; 103:4918 - 4923
- Schuler B, Furst F, Osterroth F, Steinbacher S, Huber R, Seckler R. Plasticity and steric strain in a parallel beta-helix: rational mutations in the P22 tailspike protein. Proteins 2000; 39:89 - 101
- Schuler B, Rachel R, Seckler R. Formation of fibrous aggregates from a non-native intermediate: the isolated P22 tailspike beta-helix domain. J Biol Chem 1999; 274:18589 - 18596
- Evans MS, Sander IM, Clark PL. Cotranslational folding promotes beta-helix formation and avoids aggregation in vivo. J Mol Biol 2008; 383:683 - 692
- Saa P, Castilla J, Soto C. Ultra-efficient replication of infectious prions by automated protein misfolding cyclic amplification. J Biol Chem 2006; 281:35245 - 35252