Abstract
To understand why cross species infection of prion disease often results in inefficient transmission and reduced protein conversion, most research has focussed on defining the effect of variations in PrP primary structures, including sequence compatibility of substrate and seed. By contrast, little research has been aimed at investigating structural differences between different variants of PrPC and secondary structural requirements for efficient conversion. This is despite a clear role for molecular chaperones in formation of prions in non-mammalian systems, indicating the importance of secondary/tertiary structure during the conversion process. Recent data from our laboratory on the cellular location of disease-specific prion cofactors supports the critical role of specific secondary structural motifs and the stability of these motifs in determining the efficiency of disease-specific prion protein conversion. In this paper we summarise our recent results and build on the hypothesis previously suggested by Wuthrich and colleagues, that stability of certain regions of the prion protein is crucial for protein conversion to abnormal isoforms in vivo. It is suggested that one role for molecular co-factors in the conversion process is to stabilise PrPC structure in a form that is amenable for conversion to PrPSc.
Introduction
Prion diseases in mammals, or transmissible spongiform encephalopathies (TSEs), exist as a family of different strains that appear to be encoded by distinct conformationsCitation1 of the disease-associated form of the prion protein, PrPSc. How a single, rather small protein can fold into multiple discrete structures that are all capable of causing disease remains puzzling. It has been hypothesized that strain-specific cofactors exist that are capable of catalyzing the formation of PrPSc-like conformationsCitation2–Citation4 from the native isoform, PrPC. Indirect evidence for the existence of cofactors comes from the repeated failure to refold purified recombinant PrP (recPrP) into a structure that recapitulates the infectivity levels associated with PrPSc or replicates all characteristics of a natural TSE disease.Citation5–Citation8 However, the Supattapone laboratory has recently published studies demonstrating that purified PrPC can be refolded, in the presence of lipid, nucleic acid and a seed of PrPSc, into forms that are fully infectious.Citation9–Citation11 Earlier this year, these studies culminated when Wang et al.Citation12 published results that appear to show the generation of prion infectivity from recombinant PrP and purified components in the absence of PrPSc. Many of these recent studies use the protein misfolding cyclic amplification (PMCA) technologyCitation13 to induce misfolding. This technique incorporates sequential sonication steps, presumably to break up aggregates and propagate greater quantities of smaller seeds.Citation14 However, there are a range of problems associated with the use of sonication to aid protein misfolding; sonication is clearly non-physiological and the energy imparted may overcome the absence of as yet undiscovered cofactors. The lack of quantitative data from PMCA applications does not allow certain mechanistic aspects of protein misfolding to be investigated and sonication has also been shown to generate amyloid-like fibrils from proteins not known to fibrillize in vivo.Citation15,Citation16 Even in the absence of known prion cofactors or understanding of the role of sonication in prion amplification, it is critically important to investigate prion misfolding mechanisms under physiological conditions.
We have traditionally used a modified versionCitation17 of the cell-free conversion assay (CFCA), a technique pioneered during the 1990s by the group of Byron Caughey.Citation18 This assay is a physiological prion conversion system that mimics many aspects of TSE disease, including species barriers and the effect of polymorphisms. Conversion of recombinant PrP in the assay is also inhibited by known anti-prion drugs.Citation19 By use of the CFCA, we have recently published two studiesCitation20,Citation21 that appear unrelated, but which taken together suggest that partially stabilizing the structure of PrPC in a form that is amenable for conversion to PrPSc is a potential role for prion cofactors. Our data agree with and extend previous observations on the balance between different misfolding pathways of PrP and the role of key areas of secondary structure of PrPC in directing conversion.
Specific Fractions of Scrapie-Susceptible Cells Enhance Disease-Specific PrP Misfolding but Inhibit Generic Misfolding
In work published in the Journal of Biological Chemistry,Citation20 we described experiments by which we investigated the location of prion conversion cofactors through fractionation of scrapie-susceptible cells and use of the subcellular fractions to supplement our cell-free conversion assay. The CFCA involves the incubation of recPrP with a seed of PrPSc enriched from mouse brains and, after proteinase K (PK) digestion, we can measure the amount of recPrP that has converted to a protease-resistant isoform, following which the percent efficiency of conversion can be assessed.Citation17 We found several subcellular fractions that had little or no effect on conversion efficiency when added to the assay while other fractions resulted in degradation of the recombinant PrP substrate, which demonstrates the presence of proteases in those preparations. Crucially, however, we found some subcellular fractions that double the efficiency of conversion of recombinant PrP compared to control reactions. Subcellular fractions were prepared by ultracentrifugation through a gradient of OptiPrep and fractions enhancing PrP conversion were from the top of the gradient, representing low density species. We showed that these low density fractions were composed of proteins from both the cell membrane and the cytoplasm and enhancement of PrP conversion proceeded in a dose-dependent manner. Thus, the moieties in these fractions enhance cell-free conversion and are putative prion misfolding cofactors.
In order to test specificity of these results, we used a conversion system seeded with PrPSc derived from mice infected with ME7 scrapie and demonstrated enhanced conversion in the presence of low density fractions from both ME7-susceptible LD9 cells and ME7 non-susceptible SMB cells.Citation22 This result was surprising since SMB cells are not susceptible to stable infection with ME7 scrapie (Ruth Hennion, personal communication), a phenomenon that has been suggested to result from the lack of strain-specific cofactors.Citation1 A similar result was achieved using low density fractions from LD9 cells and a conversion assay system seeded with PrPSc from the 79A strain of scrapie, a strain that has not been shown to infect LD9 cells.Citation3 Thus, we concluded that the effects of the subcellular fractions on conversion were at least partly non-specific. In order to investigate the mechanism of conversion enhancement, we also tested the effect of these subcellular fractions on an alternative misfolding pathway of PrP: denaturant- and shaking-induced fibrillization.Citation23 Surprisingly, the same low density, subcellular fractions that enhanced disease-specific misfolding in the CFCA inhibited fibrillization of recombinant murine PrP. In more recent work, we have also tested the species specificity of subcellular fractions by fibrillizing ovine recombinant PrP in the presence of the fractions (derived from the murine LD9 cell line). The results are summarized in , which shows fibrillization curves of ovine recombinant PrP in the presence of fractions 1, 2 and 10, as monitored by thioflavin T (ThT) fluorescence, together with a bar chart of lag times of all fibrillization reactions. These data essentially replicate our previous findings using murine recombinant PrP; low density fractions that enhance cell-free conversion (fractions 2–5) also inhibit fibrillization, in this case across an ovine-murine species barrier, further demonstrating the lack of specificity of the cofactors present in the subcellular fractions.
If the cofactors present in our low density subcellular fractions act non-specifically, both in terms of TSE strain and species, to enhance disease-specific prion protein misfolding and inhibit in vitro fibrillization, then what is the mechanism of action? Furthermore, how are species barriers mediated and what limits the ability of different scrapie strains to infect particular cells and/or individuals? Traditional dogma suggests that compatibility between exogenous PrPSc and host PrPC is an important factor in mediating species barriersCitation24 but is not sufficient to explain all aspects of TSE infections. Equally, cell-specific strain tropism is believed to result from restricted expression of cellular cofactors,Citation1 but our data argues against this. Differential clearance of PrPSc from TSE-infected cells may also play a key role in suppressing certain infections, be that different TSE strains or infections passed from other species. Certainly, our data, albeit using in vitro systems, suggest that cell-derived, low density cofactors may help replicate all forms of TSE disease and that some mechanisms limiting infection may operate on a cellular rather than molecular level.
Suggestions of how our cell-derived cofactor preparations may function can be derived by considering details of the two different conversion systems used. The CFCA uses physiological salts supplemented with detergent and a seed of PrPSc to convert recombinant PrP. Basic research into the mechanism of this assay suggests that the correct fold of the substrate is essential,Citation25 although sub-denaturing concentrations of guandinium hydrochloride can also enhance the assay efficiency, presumably by “loosening” the structure of PrPSc. Conversely, the fibrillization experiment, as pioneered by Baskakov et al.Citation23 proceeds under more denaturing conditions and the results of various experiments suggest that at least partial unfolding of the recPrP to a prefibrillar intermediate is an important step in this assay.Citation26,Citation27 For example, shows the effect of reduced guanidine concentrations on fibrillization of murine PrP; as the concentration of guanidine is reduced from 2 to 0.24 M the lag time to fibrillization increases dramatically in both unseeded assays and those seeded with preformed fibrils. Thus, it is clear that partially denaturing the structure of recombinant PrP favors formation of non-disease-specific isoforms, while maintaining and possibly stabilizing the structure of recombinant PrP aids formation of disease-specific structures. A basic schematic depicting this balance is shown in . Solution conditions or molecular cofactors could, therefore, tip the balance of the “folded ↔ partially folded” equilibrium in one direction or the other. If true, this finding has implications for therapeutics that seek to stabilize the structure of PrPC; they may inadvertently stabilize PrPC in a structure more amenable for disease-specific conversion.
An Ovine-Specific Mutation Reduces Stability of Murine Recombinant PrP but Also Reduces Efficiency of Conversion
We have also recently published work in BiochemistryCitation21 that indirectly supports the hypothesis that a well-folded and stable structure for PrPC is critically important for efficient conversion to PrPSc. This paper describes experiments in which we investigated the effect of a specific polymorphism in ovine PrP and the data obtained may point, more generally, to principles underlying PrP structure and conversion. The ovine prnp gene is highly polymorphic and a number of polymorphisms have been shown to have profound effects on the susceptibility of sheep to both natural and experimental scrapie.Citation28 One such polymorphism, discovered recently by colleagues at The Roslin Institute, involves the amino acid substitution Pro-Leu at codon 168 (P168L) and the mutation confers dramatically increased resistance of sheep to experimental BSE infection.Citation29 We previously showed that recombinant ovine PrP carrying the P168L substitution was resistant to conversion in vitro and, furthermore, that murine PrP carrying the equivalent substitution (P164L) was also resistant to conversion.Citation30 To investigate the mechanism of this polymorphism-mediated resistance to conversion (and disease) we expressed a range of murine proteins carrying different substitutions at codon 164 (164P, 164L, 164Q, 164E and 164S) and studied the thermal stability of the proteins and their ability to convert to a protease-resistant isoform in the CFCA. We found a correlation between stability and extent of conversion, such that the two most thermally stable proteins (164P and 164S) were converted the most efficiently, while the other three variants had reduced stability and were converted less efficiently. Consistent with our hypothesis that reduced stability promotes non-disease-specific misfolding, we further found that in vitro oligomerization of the 164L variant, which converts inefficiently in the CFCA and is thermally less stable, was more rapid than the 164P variant, which converts efficiently in the CFCA and is thermally more stable. Oligomerization of recombinant PrP was carried out under partly denaturing conditions at acidic pH,Citation31 thus our data is consistent with an increased tendency of unstable proteins to adopt intermediates on the pathway to generically misfolded isoforms. These observations agree with the basic schematic depicted in .
Other than codon 164 amino acid changes, the destabilizing effects of other amino acid substitutions on PrP structure have been published, most notably ovine-specific polymorphisms.Citation32–Citation34 These studies indicate that the substitution Gln-Arg at codon 171, which is close to residue 168 and is associated with extreme resistance of sheep to classical scrapie, produces a protein with reduced stability. This effect has been used to posit the hypothesis that such proteins misfold generically in vivo more readily and hence are cleared more rapidly leading to reduced protein half-life and reduced potential substrate for conversion. It is not facile to generate directly comparable data when measuring differences in protein turnover rates as a function of amino acid changes and this hypothesis remains controversial. However, our data would suggest that increased turnover of “resistant” proteins need not be the only mechanism by which disease resistance is modulated. Instead, we note that less stable protein variants convert less readily in the CFCA, a system in which increased protein turnover cannot be invoked as an argument.Citation30 This leads to the conclusion that a stable and well-folded substrate is a prerequisite for efficient conversion, a conclusion that correlates directly with our proposed mechanism for the enhancement of conversion by our low density subcellular fractions.
Further evidence of the role of stability in this region of PrP comes from studies of the molecular differences between cervid and murine PrP. Cervids appear exceptionally susceptible to chronic wasting disease (CWD) and it has always been puzzling why the disease has spread so readily in elk and deer in the US. Part of this appears to involve the ready secretion of CWD infectivity from deer,Citation35 but following determination of the NMR structure of elk prion protein by the group of Wuthrich, another possible reason for the apparent susceptibility of deer became apparent.Citation36 The deer prion protein contains a well defined loop region connecting the second β-sheet to the second α-helix. In most other species studied, this loop is at least partially mobile and Gossert et al. argued that a “rigid loop” may correlate with prion disease susceptibility. Support for this comes from the finding that bank vole PrP also contains a rigid β2-α2 loopCitation37—bank voles are highly susceptible to many strains of TSE diseaseCitation38 and the reasons for this have been tracked to specific amino acid residues in the loop region of bank vole PrP that affect protein misfolding both in vitroCitation39 and in vivo.Citation40 As previously discussed, the β2-α2 loop contains the polymorphic ovine residues 168 and 171, key determinants of the susceptibility of sheep to prion disease and mediators of protein stability.
If a rigid β2-α2 loop is responsible for increased susceptibility of cervids to TSE disease then engineering such a loop in the prion protein of an alternate species should render that species more susceptible than wildtype counterparts. Sigurdson et al. created transgenic mice expressing PrP in which two endogenous murine amino acids of the loop region had been replaced by cervid counterpartsCitation41 at codons 169 and 173. These mice, therefore, expressed “rigid loop” PrP and the mice acquired a spontaneous neurodegenerative disease that was transmissible. The mice are also differentially susceptible to prion diseases from a variety of species and susceptibility was shown to be determined by compatibility between the amino acid at codon 169 in inoculum (PrPSc) and host (PrPC) prion proteins.Citation42 At a protein level, we have recently confirmed that the doubly-mutated murine protein (S169N, N173T) is thermally more stable than wildtype mouse PrP and fibrillizes less readily (Agarwal et al. in preparation) consistent with out proposed misfolding pathway in . It is also notable that this is the region of the protein that was suggested to bind to the putative cofactor protein X.Citation43 The involvement of a proteinaceous cofactor was invoked to explain the differential susceptibility of transgenic mice expressing various mouse/human chimeric PrP molecules. However, a similar experiment involving mouse/cattle chimeric proteinCitation44 failed to replicate the previous results, casting the involvement of protein X into doubt. While it remains to be tested, it appears plausible that the different protein variants involved in the studies have different levels of stability around the β2-α2 loop region, possibly through disruption of the native salt bridgesCitation45 in PrPC, and that no other protein was involved. The recent finding that the effects of chimeric murine/human prion proteins are replicable in purified systems in vitro adds weight to this argument.Citation46 Thus, several lines of evidence support the hypothesis that stability in PrPC and, in particular the β2-α2 loop region, is a critical determinant for disease-specific prion misfolding.
While there exists significant evidence supporting the importance of stability, there are also elements of TSE science that argue against prion protein stability as a general factor in determining disease susceptibility. Firstly, many disease-associated mutations in the human prion gene appear to reduce protein stability, apparently in direct disagreement with our hypothesis. However, the diseases that result from prion mutations do so without apparent exogenous infection and manifest only late in life, suggesting that the proteins involved may have compromised conversion efficiency once the misfolding cascade has started. Other disease-causing mutations have little effect on stability or are slightly stabilizing and despite much research (reviewed in ref. Citation47) mechanistic aspects of these mutations remain to be established. Secondly, the recently identified atypical scrapie has a pattern of susceptibility that is essentially the reverse of that known for classical scrapie, such that sheep expressing proteins that are less stable are more susceptible to disease.Citation48 While atypical scrapie has been shown to be transmissible,Citation49 the small number of cases encountered and late age of onset is more suggestive of a sporadic disease and in such cases a less stable protein may be critical to initiate the infection. The structure of the PrPSc deposited during pathogenesis of atypical scrapie also appears different to that formed during classical scrapie, as evidenced by the presence of a low molecular weight isoform after proteinase K digestion.Citation50 This is somewhat more reminiscent of the PK-resistant cores of certain forms of sporadic CJD and fibrils of recombinant PrP.Citation51 As more research in this area is published it may become evident that protein stability is important only for certain TSE strains. It is also possible that, while stability in the loop region is important for efficient conversion, reduced stability in other regions may also play a role in mediating conversion. Due to loss of key hydrogen bonds or salt bridges, it is possible that single amino acid substitutions can have stabilizing effects in some parts of the molecules but destabilizing effects in others. Hence, the importance of secondary structural stability in protein conversion may be dependent on the particular amino acid substitution, prion protein structural motif or TSE strain in question.
What are the In Vivo Cofactors that Enhance Prion Conversion in Our Low Density Subcellular Fractions?
Although our combined data are consistent with alterations in conversion efficiency resulting from the ability of low density subcellular fractions to modulate protein folding and stability, we do not yet know the identities of the conversion enhancing cofactors. By use of what knowledge we have about our low density fractions and comparisons to recent literature we can speculate on the presence of particular molecules or molecular subsets that may increase PrPC stability. In our previous work we probed subcellular fractions for markers of specific organelles by western blotting and found that active fractions contained the plasma membrane marker annexin II. By use of proteomic methods, we also found a large number of cytoplasmic proteins in low density subcellular fractionsCitation20 and certain aspects of PrP cell biology could allow interactions with molecules in either location. After expression in the ER, PrPC is secreted and is anchored on the cell membrane by means of its glycosylphosphatidyl inositol membrane anchor. Here PrPC has been shown to be localized to detergent-resistant membranes (lipid rafts), but then leaves these to be endocytosed and recycled. Thus, interaction with other molecules on the plasma membrane is not only likely to allow PrPC to carry out its normal function, but interaction with a receptor is an obligatory step in endocytosis of a molecule without cytoplasmic domains. Likewise, although PrPC is probably not localized to the cytoplasm during its normal cell biology, cytoplasmic PrP has been detected after proteasome inhibitionCitation52 and these are most likely aberrantly folded PrP isoforms, formed in the ER/golgi body during normal post-translational folding and targeted for proteasomal degradation. Either cellular site, therefore, represents a plausible location for a prion conversion cofactor, with recent data from the Soto laboratory suggesting that prion cofactors may reside predominately on the plasma membrane.Citation4
If prion cofactors are either cytoplasmic or located on or around the plasma membrane, then do any known binding partners fit this expression profile? Alternatively, can molecules used in other misfolding studies be identified that may represent our conversion-enhancing cofactors? Putative prion protein interaction partners have been identified in an increasing number of studies and include, amongst others, N-CAM,Citation53 37–67 kDa laminin receptor precursor,Citation54 LRP-1,Citation55 glypican-1,Citation56 glycosaminoglycans and proteoglycansCitation57 (including heparin and chondroitin sulphates), nucleic acids,Citation58 anionic lipidsCitation59 and copper ions.Citation60 In short, almost all subsets of biomolecules are represented, many of which can be found in the extracellular milieu, on the plasma membrane or in the cytoplasm, and the result is a minefield of information, with many molecules representing possible cofactors but few with functional data in this regard. The interactome of PrPC is therefore not particularly useful for identifying putative cofactors,Citation61,Citation62 but a range of other published studies provide more specific cofactors that may be involved in altering PrPC stability.
Identification of molecules co-purifying with PrPSc in scrapie-associated fibrils.
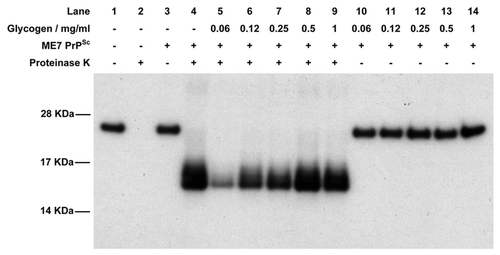
One of the more obvious places to look for prion cofactors is in the PrPSc-containing plaques and fibrils that are deposited during TSE disease. If cofactors are involved in the PrPC-PrPSc misfolding process then it seems reasonable that they should become integrated into the growing fibril. Various molecules have been suggested to co-purify with PrPSc in scrapie-associated fibrils (SAF) including polysaccharide (which may act as a scaffold), small nucleic acids (see below) and other proteins. The polysaccharide has been suggested to consist of α-linked polyglucose, similar to glycogen,Citation63 and purified glycogen was shown to promote fibrillization of PrP in vitro.Citation64 We have also tested glycogen in the cell-free conversion assay for its ability to enhance PrPSc catalyzed misfolding. Typical results are shown in and show clearly that, at low concentrations, glycogen inhibits disease-specific misfolding of PrP, although this effect is diminished at higher concentrations. This is a curious result and suggests that the effect of glycogen is related to its concentration, perhaps through formation of glycogen multimers at higher concentration that lose inhibitory activity. In any case, based on the previous fibrillization data, inhibition of cell-free conversion at low glycogen concentration is surprising, but suggests that glycogen may not act as a prion conversion cofactor. The opposite effects of glycogen on in vitro fibrillization and cell-free conversion, however, agree with our model of PrP misfolded presented in and suggest that glycogen may act, at low concentrations, to destabilize recombinant PrP structure.
By use of proteomic techniques, several groups have identified different proteins co-purifying with PrPSc in SAF preparations.Citation65–Citation67 Co-purifying proteins include ferritin, CaMKII and various cytoskeletal and structural proteins including actin and tubulin isoforms. However, some previous studies omitted control preparations from either uninfected or PrP-null mice in proteomic identifications; we have found that some of the proteins previously suggested to be specific to SAF preparations are also purified after performing mock SAF preparations on control brains, a finding confirmed by Moore et al.Citation67 This presumably results from the large size and general insolubility of these proteins. Ferritin, for example, exists as a multimer of molecular weight ∼0.5 MDa but can form higher molecular weight oligomers. We have compiled lists of proteins that are present specifically in SAF preparations after subtraction of those proteins co-purifying simply because of their biophysical properties (Graham et al. in preparation). Within the refined list are several proteins that are usually resident on the plasma membrane, cytoskeletal proteins and some cytoplasmic proteins. We are currently trialing these proteins for their ability to enhance prion protein misfolding in the cell-free conversion assay and their effects on stability of the structure of recombinant PrP.
Molecules used in misfolding studies to generate de novo infectivity.
Initial studies reporting the generation of prion infectivity de novo from purified components revealed the role for both lipids and specific nucleic acids (poly(A)RNA) in enhancing conversion of PrP. However, recent data from use of PMCA suggest that the requirement for nucleic acid for efficient prion amplification may be species-dependent; cofactors for propagation of murine and bank vole PrPSc were degraded by proteases indicating that they are at least partially proteinaceous in nature.Citation68 It is also worth noting that nucleic acid can be replaced by similarly charged glycosaminoglycans in hamster PMCA systemsCitation9 indicating that the mode of action for such cofactors may be related to charge and, on this basis, they may be playing a biophysical structural role as opposed to a biological functional role. Glycosaminoglycans have been shown to stimulate cell-free conversion of recombinant PrPCitation69 although their effect on the structural stability of the protein is not known and the mechanism by which both CFCA and PMCA assays are enhanced by polyanionic substances is not clear. Likewise, although the interaction of PrP with lipids has been studied extensively,Citation70 the mechanism of its apparent role as a cofactor in PrP amplification studies is not known and may simply bring together PrPC and PrPSc in vesicles to facilitate conversion. It remains possible that our low density fractions that enhance cell-free conversion contain both lipids and polyanionic species and the mechanism of conversion enhancement is similar to that described in PMCA studies.
An interesting variation to the constituents used for PMCA studies has been reported by Murayama et al.Citation71 In their studies they found that the spontaneous formation of non-disease-specific protease-resistant PrP could be inhibited by the addition of digitonin, a glycoside that aids solubilization of lipids and membrane proteins. Digitonin consists of a steroid covalently linked to a small sugar moiety and the stabilizing effect of small saccharides, such as trehalose,Citation72 on protein structure is well known. It is likely that digitonin helps to maintain the structural stability of the PrP substrate during PMCA applications, thereby inhibiting non-specific fibrillization and promoting disease-specific misfolding.
Heat shock proteins as potential cofactors in mammalian systems.
In yeast, a system that is genetically tractable, the importance of heat shock proteins (HSPs) in propagation of the prion states is well established,Citation73 although the exact role of each HSP is still the subject of much research. HSPs can function by ensuring that individual proteins are correctly folded but also appear to help in disaggregating misfolded protein aggregates. It has been suggested continually that HSPs are important in mammalian prion diseases and various members of the HSP family have been found to bind PrPC,Citation74 copurify with PrPSc fibrilsCitation65 or aid the conversion process in vitro.Citation75 We know that cytoplasmic HSPs are present in our low density subcellular fractions since Hsp90 isoforms were detected by proteomic methods. They may represent the cofactors that enhance cell-free conversion by ensuring a correctly folded substrate and may act similarly to inhibit in vitro fibrillization of recombinant PrP. Whether cytoplasmic HSPs interact with PrPC in vivo is not clear, although retrograde transport of PrPC from the endoplasmic reticulum to the cytoplasm provides a mechanism by which this interaction may occur.Citation52
Experimental Procedures
All experimental procedures used to generate data shown in this paper have been previously published.Citation20,Citation21
Conclusion
Contrary to expectations, resistance-associated PrPC proteins appear less stable than proteins associated with disease susceptibility, highlighting the possibility that a stable PrP molecule, in particular the β2-α2 loop, may be a prerequisite for efficient PrP conversion in acquired prion diseases. Our recent data supports this hypothesis through the identification of PrP loop mutants that correlates stability with in vitro conversion and through the identification of low density subcellular fractions that enhance cell-free conversion but inhibit fibrillization. To fully understand implications of both findings for TSE disease it will be important to define completely the role of PrP structure in the conversion process.
Abbreviations
| CFCA | = | cell-free conversion assay |
| CWD | = | chronic wasting disease |
| HSP | = | heat shock protein |
| PK | = | proteinase K |
| PMCA | = | protein misfolding cyclic amplification |
| PrPC | = | cellular prion protein |
| PrPSc | = | disease-associated prion protein |
| recPrP | = | recombinant PrP |
| SAF | = | scrapie-associated fibrils |
| TSE | = | transmissible spongiform encephalopathy |
Figures and Tables
Figure 1 Fibrilization of ovine PrP, monitored by thioflavin T fluorescence, in the presence of subcellular fractions of murine LD9 cells. (A) Fraction 1 (B) Fraction 2 (C) Fraction 10 (D) Bar chart showing lag times to fibrilization of ovine PrP in the presence of all 22 fractions produced by fitting sigmoidal curves to raw ThT fluorescence data. Reactions supplemented with fractions 2–5 did not fibrilize completely in the time frame of the assay (24 h) hence sigmoidal cures could not accurately be fitted to allow lag times to be calculated. The same fractions (2–5) were previously shown to enhance conversion of PrP to a protease-resistant isoform in the cell-free conversion assay. The +ve control relates to fibrilization reactions to which no subcellular fractions were added.
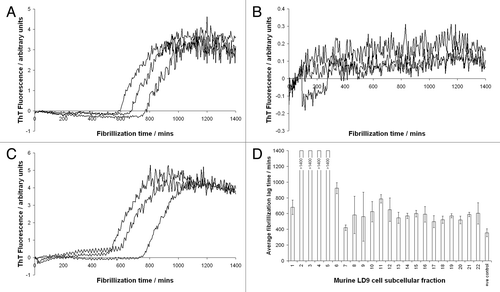
Figure 2 The effect of guanidinium hydrochloride concentration on fibrillization of murine PrP. Guanidine in the final reaction was titrated and lag times to fibrillization of either unseeded (white bars) or seeded (filled bars) reactions were calculated by fitting sigmoidal curves to raw ThT fluorescence data. For unseeded reactions at low guanidine concentrations the protein did not fibrillize to completion in the timeframe of the assay (24 h) hence lag times could not be calculated.
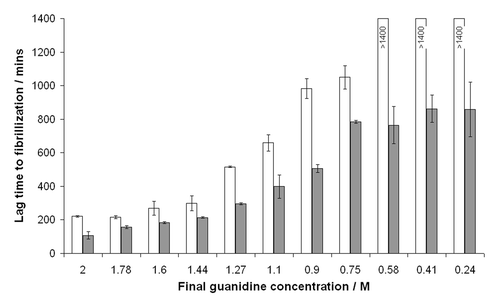
Figure 3 Schematic of a proposed, simplified pathway of PrP folding. Partial denaturation to an intermediate conformation promotes generic misfolding to oligomers and fibrils. Disease-specific misfolding proceeds from a more fully folded form, possibly by route of a different, PrPSc-induced, partially folded intermediate. General conditions that promote unfolding will lead more rapidly to generic misfolded isoforms, while those that promote stability and a more structured PrPC molecule will favor disease-specific misfolding.
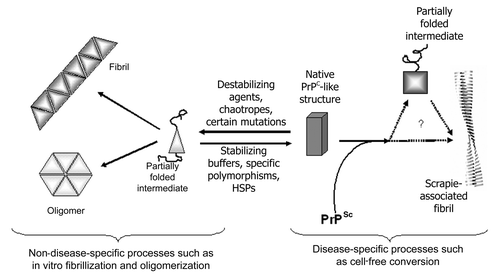
Figure 4 Western blot of the cell-free conversion assay of 3F4 antibody-tagged murine recombinant PrP in the presence of glycogen. Assays were seeded with PrPSc purified from ME7-infected mice and, in each case, 9/10th of the reaction was treated with PK while the remaining 1/10th was not. In the absence of PrPSc, recombinant PrP is digested away by PK (lanes 1 and 2) while in the presence of PrPSc a protease-resistant isoform is produced (lanes 3 and 4). In the presence of low concentrations of glycogen, cell-free conversion is inhibited (lanes 5–7) while at higher concentrations of glycogen conversion is similar to control levels (lanes 9 and 10). Lanes 11–14 contain the non-PK treated samples in the presence of glycogen to demonstrate that similar levels of recPrP substrate were present in each assay.
Acknowledgements
We gratefully acknowledge Prof Charles Weissmann for provision of LD9 cells used in some studies, Dr. Louise Kirby and Miss Pooja Krishnaswamy for molecular biology and Dr. Sandra McCutcheon for critical review of the manuscript. Work discussed in this paper was funded by BBSRC, UK.
References
- Weissmann C. Thoughts on mammalian prion strains. Folia Neuropathol 2009; 47:5 - 8
- Fasano C, Campana V, Zurzolo C. Prions: protein only or something more? Overview of potential prion cofactors. J Mol Neurosci 2006; 29:195 - 214
- Mahal SP, Baker CA, Demczyk CA, Smith EW, Julius C, Weissmann C. Prion strain discrimination in cell culture: The cell panel assay. Proc Natl Acad Sci USA 2007; 104:20908 - 20913
- Abid K, Morales R, Soto C. Cellular factors implicated in prion replication. Febs Lett 2010; 584:2409 - 2414
- Legname G, Baskakov IV, Nguyen HO, Riesner D, Cohen FE, DeArmond SJ, et al. Synthetic mammalian prions. Science 2004; 305:673 - 676
- Makarava N, Kovacs GG, Bocharova O, Savtchenko R, Alexeeva I, Budka H, et al. Recombinant prion protein induces a new transmissible prion disease in wild-type animals. Acta Neuropathol 2010; 119:177 - 187
- Colby DW, Giles K, Legname G, Wille H, Baskakov IV, DeArmond SJ, et al. Design and construction of diverse mammalian prion strains. Proc Natl Acad Sci USA 2009; 106:20417 - 20422
- Kim JI, Cali I, Surewicz K, Kong Q, Raymond GJ, Atarashi R, et al. Mammalian prions generated from bacterially expressed prion protein in the absence of any mammalian cofactors. J Biol Chem 2010; 285:14083 - 14087
- Deleault NR, Geoghegan JC, Nishina K, Kascsak R, Williamson RA, Supattapone S. Protease-resistant prion protein amplification reconstituted with partially purified substrates and synthetic polyanions. J Biol Chem 2005; 280:26873 - 26879
- Deleault NR, Harris BT, Rees JR, Supattapone S. Formation of native prions from minimal components in vitro. Proc Natl Acad Sci USA 2007; 104:9741 - 9746
- Deleault NR, Lucassen RW, Supattapone S. RNA molecules stimulate prion protein conversion. Nature 2003; 425:717 - 720
- Wang F, Wang XH, Yuan CG, Ma JY. Generating a prion with bacterially expressed recombinant prion protein. Science 2010; 327:1132 - 1135
- Saborio GP, Permanne B, Soto C. Sensitive detection of pathological prion protein by cyclic amplification of protein misfolding. Nature 2001; 411:810 - 813
- Piening N, Weber P, Giese A, Kretzschmar H. Breakage of PrP aggregates is essential for efficient autocatalytic propagation of misfolded prion protein. Biochem Bioph Res Co 2005; 326:339 - 343
- Ohhashi Y, Kihara M, Naiki H, Goto Y. Ultrasonication-induced amyloid fibril formation of beta(2)-microglobulin. J Biol Chem 2005; 280:32843 - 32848
- Stathopulos PB, Scholz GA, Hwang YM, Rumfeldt JAO, Lepock JR, Meiering EM. Sonication of proteins causes formation of aggregates that resemble amyloid. Protein Sci 2004; 13:3017 - 3027
- Kirby L, Hope J. Lehmann S, Grassi J. Cell-free conversion of prion proteins. Techniques in Prion Research—Methods and Tools in Biosciences and Medicine 2004; Basel Birkhauser
- Kocisko DA, Come JH, Priola SA, Chesebro B, Raymond GJ, Lansbury PT, et al. Cell-free formation of protease-resistant prion protein. Nature 1994; 370:471 - 474
- Kirby L, Birkett CR, Rudyk H, Gilbert IH, Hope J. In vitro cell-free conversion of bacterial recombinant PrP to Prp(res) as a model for conversion. J Gen Virol 2003; 84:1013 - 1020
- Graham JF, Agarwal S, Kurian D, Kirby L, Pinheiro TJT, Gill AC. Low density subcellular fractions enhance disease-specific prion protein misfolding. J Biol Chem 2010; 285:9868 - 9880
- Kirby L, Agarwal S, Graham JF, Goldmann W, Gill AC. Inverse correlation of thermal lability and conversion efficiency for five prion protein polymorphic variants. Biochemistry-US 2010; 49:1448 - 1459
- Birkett CR, Hennion RM, Bembridge DA, Clarke MC, Chree A, Bruce ME, et al. Scrapie strains maintain biological phenotypes on propagation in a cell line in culture. EMBO J 2001; 20:3351 - 3358
- Breydo L, Makarava N, Baskakov IV. Methods for conversion of prion protein into amyloid fibrils. Methods Mol Biol 2008; 459:105 - 115
- Moore RA, Vorberg I, Priola SA. Species barriers in prion diseases—brief review. Arch Virol 2005; 187 - 202
- Herrmann LM, Caughey B. The importance of the disulfide bond in prion protein conversion. Neuroreport 1998; 9:2457 - 2461
- Kazlauskaite J, Young A, Gardner CE, Macpherson JV, Venien-Bryan C, Pinheiro TJT. An unusual soluble beta-turn-rich conformation of prion is involved in fibril formation and toxic to neuronal cells. Biochem Bioph Res Co 2005; 328:292 - 305
- Baskakov IV, Legname G, Gryczynski Z, Prusiner SB. The peculiar nature of unfolding of the human prion protein. Protein Sci 2004; 13:586 - 595
- Goldmann W. PrP genetics in ruminant transmissible spongiform encephalopathies. Vet Res 2008; 39
- Goldmann W, Houston F, Stewart P, Perucchini M, Foster J, Hunter N. Ovine prion protein variant A(136) R(154)L(168)Q(171) increases resistance to experimental challenge with bovine spongiform encephalopathy agent. J Gen Virol 2006; 87:3741 - 3745
- Kirby L, Goldmann W, Houston F, Gill AC, Manson JC. A novel, resistance-linked ovine PrP variant and its equivalent mouse variant modulate the in vitro cell-free conversion of rPrP to PrPres. J Gen Virol 2006; 87:3747 - 3751
- Rezaei H, Eghiaian F, Perez J, Doublet B, Choiset Y, Haertle T, et al. Sequential generation of two structurally distinct ovine prion protein soluble oligomers displaying different biochemical reactivities. J Mol Biol 2005; 347:665 - 679
- Robinson PJ, Pinheiro TJT. The unfolding of the prion protein sheds light on the mechanisms of prion susceptibility and species barrier. Biochemistry-US 2009; 48:8551 - 8558
- Fitzmaurice TJ, Burke DF, Hopkins L, Yang SJ, Yu SL, Sy MS, et al. The stability and aggregation of ovine prion protein associated with classical and atypical scrapie correlates with the ease of unwinding of helix-2. Biochem J 2008; 409:367 - 375
- Bujdoso R, Burke DF, Thackray AM. Structural differences between allelic variants of the ovine prion protein revealed by molecular dynamics simulations. Proteins 2005; 61:840 - 849
- Mathiason CK, Powers JG, Dahmes SJ, Osborn DA, Miller KV, Warren RJ, et al. Infectious prions in the saliva and blood of deer with chronic wasting disease. Science 2006; 314:133 - 136
- Gossert AD, Bonjour S, Lysek DA, Fiorito F, Wuthrich K. Prion protein NMR structures of elk and of mouse/elk hybrids. Proc Natl Acad Sci USA 2005; 102:646 - 650
- Christen B, Perez DR, Hornemann S, Wuthrich K. NMR structure of the bank vole prion protein at 20 degrees C contains a structured loop of residues 165–171. J Mol Biol 2008; 383:306 - 312
- Di Bari MA, Chianini F, Vaccari G, Esposito E, Conte M, Eaton SL, et al. The bank vole (Myodes glareolus) as a sensitive bioassay for sheep scrapie. J Gen Virol 2008; 89:2975 - 2985
- Piening N, Nonno R, Di Bari M, Walter S, Windl O, Agrimi U, et al. Conversion efficiency of bank vole prion protein in vitro is determined by residues 155 and 170, but does not correlate with the high susceptibility of bank voles to sheep scrapie in vivo. J Biol Chem 2006; 281:9373 - 9384
- Agrimi U, Nonno R, Dell'Omo G, Di Bari MA, Conte M, Chiappini B, et al. Prion protein amino acid determinants of differential susceptibility and molecular feature of prion strains in mice and voles. PloS Patho 2008; 4:1000113
- Sigurdson CJ, Nilsson KP, Hornemann S, Heikenwalder M, Manco G, Schwarz P, et al. De novo generation of a transmissible spongiform encephalopathy by mouse transgenesis. Proc Natl Acad Sci USA 2009; 106:304 - 309
- Sigurdson CJ, Nilsson KP, Hornemann S, Manco G, Fernandez-Borges N, Schwarz P, et al. A molecular switch controls interspecies prion disease transmission in mice. J Clin Invest 2010; 120:2590 - 2599
- Kaneko K, Zulianello L, Scott M, Cooper CM, Wallace AC, James TL, et al. Evidence for protein X binding to a discontinuous epitope on the cellular prion protein during scrapie prion propagation. Proc Natl Acad Sci USA 1997; 94:10069 - 10074
- Scott MR, Safar J, Telling G, Nguyen O, Groth D, Torchia M, et al. Identification of a prion protein epitope modulating transmission of bovine spongiform encephalopathy prions to transgenic mice. Proc Natl Acad Sci USA 1997; 94:14279 - 14284
- Guest WC, Cashman NR, Plotkin SS. Electrostatics in the stability and misfolding of the prion protein: salt bridges, self energy and solvation. Biochem Cell Biol 2010; 88:371 - 381
- Geoghegan JC, Miller MB, Kwak AH, Harris BT, Supattapone S. Trans-dominant inhibition of prion propagation in vitro is not mediated by an accessory cofactor. PLoS Pathog 2009; 5:1000535
- van der Kamp MW, Daggett V. The consequences of pathogenic mutations to the human prion protein. Protein Eng Des Sel 2009; 22:461 - 468
- Luhken G, Buschmann A, Brandt H, Eiden M, Groschup MH, Erhardt G. Epidemiological and genetical differences between classical and atypical scrapie cases. Vet Res 2007; 38:65 - 80
- Simmons MM, Konold T, Simmons HA, Spencer YI, Lockey R, Spiropoulos J, et al. Experimental transmission of atypical scrapie to sheep. BMC Vet Res 2007; 3:20
- Foster J, Toovey L, McKenzie C, Chong A, Parnham D, Drummond D, et al. Atypical scrapie in a sheep in a closed UK flock with endemic classical natural scrapie. Vet Rec 2008; 162:723 - 724
- Bocharova OV, Breydo L, Salnikov VV, Gill AC, Baskakov IV. Synthetic prions generated in vitro are similar to a newly identified subpopulation of PrPSc from sporadic Creutzfeldt-Jakob disease. Protein Sci 2005; 14:1222 - 1232
- Ma J, Lindquist S. Wild-type PrP and a mutant associated with prion disease are subject to retrograde transport and proteasome degradation. Proc Natl Acad Sci USA 2001; 98:14955 - 14960
- Schmitt-Ulms G, Legname G, Baldwin MA, Ball HL, Bradon N, Bosque PJ, et al. Binding of neural cell adhesion molecules (N-CAMs) to the cellular prion protein. J Mol Biol 2001; 314:1209 - 1225
- Rieger R, Edenhofer F, Lasmezas CI, Weiss S. The human 37 kDa laminin receptor precursor interacts with the prion protein in eukaryotic cells. Nature Med 1997; 3:1383 - 1388
- Parkyn CJ, Vermeulen EG, Mootoosamy RC, Sunyach C, Jacobsen C, Oxvig C, et al. LRP1 controls biosynthetic and endocytic trafficking of neuronal prion protein. J Cell Sci 2008; 121:773 - 783
- Taylor DR, Whitehouse IJ, Hooper NM. Glypican-1 mediates both prion protein lipid raft association and disease isoform formation. PLoS Pathog 2009; 5:e1000666
- Brimacombe DB, Bennett AD, Wusteman FS, Gill AC, Dann JC, Bostock CJ. Characterization and polyanion-binding properties of purified recombinant prion protein. Biochem J 1999; 342:605 - 613
- Gabus C, Auxilien S, Pechoux C, Dormont D, Swietnicki W, Morillas M, et al. The prion protein has DNA strand transfer properties similar to retroviral nucleocapsid protein. J Mol Biol 2001; 307:1011 - 1021
- Sanghera N, Pinheiro TJ. Binding of prion protein to lipid membranes and implications for prion conversion. J Mol Biol 2002; 315:1241 - 1256
- Brown DR, Qin K, Herms JW, Madlung A, Manson J, Strome R, et al. The cellular prion protein binds copper in vivo. Nature 1997; 390:684 - 687
- Rutishauser D, Mertz KD, Moos R, Brunner E, Rulicke T, Calella AM, et al. The comprehensive native interactome of a fully functional tagged prion protein. PLoS One 2009; 4:4446
- Nieznanski K. Interactions of prion protein with intracellular proteins: so many partners and no consequences?. Cell Mol Neurobiol 2010; 30:653 - 666
- Dumpitak C, Beekes M, Weinmann N, Metzger S, Winklhofer KF, Tatzelt J, et al. The polysaccharide scaffold of PrP 27–30 is a common compound of natural prions and consists of alpha-linked polyglucose. Biol Chem 2005; 386:1149 - 1155
- Panza G, Stohr J, Birkmann E, Riesner D, Willbold D, Baba O, et al. Aggregation and amyloid fibril formation of the prion protein is accelerated in the presence of glycogen. Rejuvenation Res 2008; 11:365 - 369
- Giorgi A, Di Francesco L, Principe S, Mignogna G, Sennels L, Mancone C, et al. Proteomic profiling of PrP27–30-enriched preparations extracted from the brain of hamsters with experimental scrapie. Proteomics 2009; 9:3802 - 3814
- Petrakis S, Malinowska A, Dadlez M, Sklaviadis T. Identification of proteins co-purifying with scrapie infectivity. J Proteomics 2009; 72:690 - 694
- Moore RA, Timmes A, Wilmarth PA, Priola SA. Comparative profiling of highly enriched 22L and Chandler mouse scrapie prion protein preparations. Proteomics 2010; 10:2858 - 2869
- Deleault NR, Kascsak R, Geoghegan JC, Supattapone S. Species-dependent differences in cofactor utilization for formation of the protease-resistant prion protein in vitro. Biochemistry-US 2010; 49:3928 - 3934
- Wong C, Xiong LW, Horiuchi M, Raymond L, Wehrly K, Chesebro B, et al. Sulfated glycans and elevated temperature stimulate PrP(Sc)-dependent cell-free formation of protease-resistant prion protein. EMBO J 2001; 20:377 - 386
- Sanghera N, Swann MJ, Ronan G, Pinheiro TJ. Insight into early events in the aggregation of the prion protein on lipid membranes. Biochim Biophys Acta 2009; 1788:2245 - 2251
- Murayama Y, Yoshioka M, Yokoyama T, Iwamaru Y, Imamura M, Masujin K, et al. Efficient in vitro amplification of a mouse-adapted scrapie prion protein. Neurosci Lett 2007; 413:270 - 273
- Faria TQ, Knapp S, Ladenstein R, Macanita AL, Santos H. Protein stabilisation by compatible solutes: effect of mannosylglycerate on unfolding thermodynamics and activity of ribonuclease A. Chembiochem 2003; 4:734 - 741
- Tuite M, Stojanovski K, Ness F, Merritt G, Koloteva-Levine N. Cellular factors important for the de novo formation of yeast prions. Biochem Soc Trans 2008; 36:1083 - 1087
- Edenhofer F, Rieger R, Famulok M, Wendler W, Weiss S, Winnacker EL. Prion protein PrPc interacts with molecular chaperones of the Hsp60 family. J Virol 1996; 70:4724 - 4728
- DebBurman SK, Raymond GJ, Caughey B, Lindquist S. Chaperone-supervised conversion of prion protein to its protease-resistant form. Proc Natl Acad Sci USA 1997; 94:13938 - 13943