Abstract
The infectious agent of prion diseases is believed to be nucleic acid-free particles composed of misfolded conformational isomers of a host protein known as prion protein (PrP). Although this “protein-only” concept is generally accepted, decades of extensive research have not been able to elucidate the mechanisms by which PrP misfolding leads to neurodegeneration and infectivity. The challenges in studying prion diseases relate in part to the limitations of mammalian prion models, which include the long incubation period post-infection until symptoms develop, the high expense of maintaining mammals for extended periods, as well as safety issues. In order to develop prion models incorporating a genetically tractable simple system with a well-defined neuronal system, we generated transgenic C. elegans expressing the mouse PrP behind the pan-neuronal ric-19 promoter (Pric-19). We show here that high expression of Pric-19::PrP in C. elegans can result in altered morphology, defective mobility, and shortened lifespan. Low expression of Pric-19::PrP, however, does not cause any detectable harm. Using the dopamine neuron specific promoter Pdat-1, we also show that expression of the murine BAX, a pro-apoptotic member of the Bcl-2 family, causes dopamine neuron destruction in the nematode. However, co-expression of PrP inhibits BAX-mediated dopamine neuron degeneration, demonstrating for the first time that PrP has anti-BAX activity in living animals. Thus, these distinct PrP-transgenic C. elegans lines recapitulate a number of functional and neuropathological features of mammalian prion models, and provide an opportunity for facile identification of genetic and environmental contributors to prion-associated pathology.
Introduction
In mammals, the host-encoded prion protein can adopt various stable conformations, including the normal cellular (PrPC) and multiple misfolded conformations. Some of these misfolded conformations are amyloid aggregates capable of transmitting prion diseases in the absence of nucleic acids, and they are referred to as PrPSc.Citation1,Citation2 PrP is an evolutionarily conserved glycosylphosphatidylinositol (GPI)-anchored glycoprotein, which is broadly expressed throughout adult life, including skeletal muscle, kidney, heart, lymophocytes and the central nervous system (CNS).Citation3–Citation5 Mice devoid of PrPC were resistant to scrapie and did not accumulate PrPSc, showing that a continuous supply of PrP substrates is essential for PrPSc propagation.Citation6 Intracerebral injections of mice with bacterial produced PrP amyloids resulted in prion-like neurologic disorders, further demonstrating that prion infectivity is conferred by aberrant PrP conformations.Citation7,Citation8 While there is ample evidence supporting the protein-only hypothesis, little is known about how PrP misfolding leads to neurodegeneration.Citation9
A number of studies suggest that prion infectivity and toxicity are decoupled. Mice expressing mutant PrP lacking the GPI anchor produced infectious PrPSc and yet were clinically healthy.Citation10 Several PrP mutants could manifest prion-like illnesses, but were not transmissible.Citation11,Citation12 Moreover, some pathogenic PrP mutants do not even form detectable aggregates.Citation13 Transgenic mice overexpressing wild-type PrP by ∼10-fold were also shown to accumulate numerous PrP deposits in neurons and to cause spontaneous neurodegenerative illness in the absence of detectable PrPSc.Citation14 Thus, aggregation, neurotoxicity and infectivity appear to be distinct and separable properties of PrP.Citation15
The normal function of PrPC remains unclear.Citation9,Citation15,Citation16 Several studies suggest that PrPC has an anti-apoptotic activity important for neuroprotection: neurons prepared from PrP-null mice were more susceptible to apoptosis than PrP-expressing cells;Citation17 PrP expression was able to rescue primary cultures of human neurons from BAX expression and serum deprivation;Citation18 overexpression of wild-type PrP reversed the neuropathologic phenotypes in mice caused by several PrP deletion mutantsCitation12,Citation19 and PrP expression efficiently prevented BAX-induced cell death in yeast.Citation20,Citation21 However, the mechanism underlying the anti-apoptotic effect of PrPC is not clear and anti-apoptotic function of PrPC in a living animal has not been shown.
To overcome the limitations brought out by the complex mammalian models, the non-vertebrate organism Drosophila has been developed for prion research.Citation22–Citation24 Very recently, C. elegans as another non-vertebrate model was also reported showing that expression of an insertional mutation of PrP in mechanosensory neurons resulted in a progressive loss of response to touch.Citation25 In this study, we describe our effort to characterize transgenic C. elegans that broadly express the mouse PrP in whole neurons as well as selectively in dopamine neurons. These established C. elegans prion models mimic some functional and pathogenic characteristics found in mammals, which might facilitate the identification and characterization of molecules as well as pathways that are involved in prion-associated neuropathogenesis.
Results
Expression of mouse prion protein in C. elegans whole neurons.
To generate C. elegans lines that heterologously express mouse PrP in their whole neurons, the C. elegans plasmids Pric-19::PrP and Pric-19::GFP were co-injected into the gonads of young C. elegans adults ( and Materials and Methods). The pan-neuronal promoter, Pric-19, allows PrP expression in all C. elegans neurons and GFP co-expression serves as a selection marker for transgenic animals. We obtained five integrant lines of Pric-19::PrP, designated as cgIs51–55 [Pric-19(PrP)]. Immunoblot analysis using 3F4, a monoclonal antibody recognizing residues 109–112 of human PrP that were engineered into the mouse PrP open reading frame (ORF) of Pric-19::PrP (see Materials and Methods), showed that the steady-state levels of PrP in lines cgIs51–53 were significantly higher than that of lines cgIs54–55 (). We estimated that the steady state levels of PrP protein in cgIs51 worms were about 3-fold more than that of cgIs55 worms (S1A). Quantitative RT-PCR analysis also showed that PrP mRNA level in cgIs51 is approximately 1.7-fold higher than in cgIs55 worms (S1B).
Mouse PrP expressed in C. elegans neurons is glycosylated, glycophosphatidylinositol-anchored and presented on cell surface.
The mouse PrP ORF encodes a polypeptide of 254 amino acid residues.Citation33 Like other cell surface proteins, the nascent PrP polypeptide undergoes a series of posttranslational modifications in ER and Golgi, including signal peptide removal (1–22 residues), glycosylation and the C-terminal hydrophobic sequence cleavage at residue 231 followed by the addition of a GPI anchor. A proper processing of PrP following translation allows PrP to be targeted to plasma membrane.Citation34 PrP has two potential N-linked glycosylation sites (residues 180 and 196) and PrP molecules can exist in di-, mono- and un-glycosylated forms.Citation35 Consistent with mammalian studies, PrP expression in C. elegans neurons results in two major protein bands with molecular weight at ∼26 kDa and ∼29 kDa and a minor band at ∼31 kDa, which may correspond to the non-, mono- and di-glycosylated forms of PrP, respectively (). To determine whether these bands represent glycosylated PrP species, the protein extracts were treated with N-glycosidase F (PNGase F), an amidase that removes N-linked carbohydrate moieties. As expected for glycosylated proteins, PNGase F treatment results in the reduction of the two larger bands (29 and 31 kDa) and an increase in the 26 kDa band (). These results are consistent with mammalian PrP expression studies and indicate that PrP is likely N-glycosylated in C. elegans neurons.
To determine whether PrP is attached to the cell membrane in C. elegans through GPI-anchorage, we treated protein extracts prepared from PrP transgenic worms with phosphatidylinositol-specific phospholipase C (PIPLC). PIPLC digestion removes the lipid moiety from the GPI anchorage and release GPI-anchored PrP from the cell membrane. Upon Triton X-114 treatment and phase partitioning, the lipid-free PrP should be found in the aqueous phase instead of the detergent phase whereas the untreated PrP should be partitioned in the detergent phase.Citation21 As seen in , almost all of the PrP partitioned with the detergent phase prior to PIPLC treatment and post PIPLC treatment a significant portion of PrP molecules were released to the aqueous phase (), indicating that PrP synthesized in C. elegans neurons is attached to the cell membrane through a GPI-anchorage. In situ immunofluorescence analysis incorporating the 3F4 antibody further confirms that PrP is localized on the cell membrane of C. elegans neurons ( and Sup. Movie 1). Taken together, these studies show that the PrP produced in C. elegans neurons is properly N-glycosylated and correctly attached to the cell membrane by GPI-anchorage.
Different expression levels of PrP can lead to striking differences in morphology and neurological behaviors.
Careful examinations of the integrated Pric-19::PrP lines revealed that PrP expression did not cause any detectable harm to cgIs54 and cgIs55 worms, in agreement with the recent report that expression of wild-type PrP in mechanosensory neurons did not result in abnormal behavior.Citation25 These low expressers, cgIs54 and cgIs55, grew and developed normally (data not shown). However, the high expresser lines, cgIs51, cgIs52 and cgIs53, grew significantly slower than non-transgenic and cgIs54, cgIs55 worms. These high expressers also exhibited abnormal body morphology, short and dumpy-like, whereas the low expressers showed normal body size and morphology (). Lifespan behavioral analysis showed that the low expressers of cgIs55 had an average lifespan of 19 d, which is compatible to that of GFP (21 d) and non-transgenic control worms (23 d). However, the average lifespan of the high expressers (cgIs51 and cgIs53) was remarkably shortened, only 12.3 d and 10.8, respectively (). We also noticed that the higher expressers moved slowly and were uncoordinated with aging. It is well documented that uncoordinated animals with neurological problems thrash less in liquid buffers than wild-type C. elegans.Citation36 We thus conducted thrash assays to quantify the difference in mobility among the integrated Pric-19::PrP lines. As shown in , while the low expressers had thrash scores similar to that of non-transgenic worms, the thrash rates of high expressers were significantly reduced, indicating that PrP overproduction caused a defect in C. elegans body movement. Pharyngeal pumping rate assay and defecation cycle assay, however, showed that all integrated Pric-19::PrP lines behaved similarly to the non-transgenic worms with the exception that the cgIs53 worms exhibited shortened defecation cycles ( and E).
Since some uncoordinated phenotypes, such as slow movement and paralysis, can be caused by defective muscle structures,Citation37 we thus examined the muscle architecture of both low and high expressers by phalloidin staining of F-actins. All worms showed normal, organized muscle architecture, confirming that the abnormalities in behavior and morphology in high expressers were not caused by defections in F-actin assembly or muscle architecture (S2).
Murine BAX results in cellular destruction in C. elegans dopamine neurons.
The establishment of Pric-19::PrP transgenic C. elegans lines provided us an opportunity to investigate whether PrPC has an anti-apoptotic function in living worms. We chose C. elegans' well-defined dopamine neuron system to do this test. We created transgenic worms, Pdat-1::BAX, which express the murine BAX, a protein of the Bcl-2 gene family that promotes apoptosis, under the control of a dopamine neuron-specific promoter, Pdat-1.Citation29 Dopamine neurons are required for normal food-sensing behavior in C. elegans.Citation38 In the absence of food, animals move rapidly but in the presence of food, they move more slowly.Citation32 If ectopic BAX expression is able to induce cell death or destruct dopamine neurons, Pdat-1::BAX worms should result in loss or reduction of food sensing ability. As shown in , non-transgenic worms (N2) exhibited an ∼15% decrease in bending frequency upon exposure to bacteria whereas two integrant lines of Pdat-1::BAX, cgIs56–57[Pdat-1(Bax)], showed no differences in bending frequency in the presence and absence of food, indicating that the function of dopamine neurons of Pdat-1::BAX worms was compromised due to BAX expression.
PrP protects dopamine neurons from BAX-mediated destruction.
We next examined whether PrP expression was able to inhibit BAX-induced cellular destruction. We crossed cgIs55, the low expresser line of Pric-19::PrP, with cgIs56, an integrated line of Pdat-1::BAX. Progeny co-expressing PrP and BAX were selected and assayed for their food sensing behaviors. As shown in , worms co-expressing PrP and BAX had a food sensing ability comparable with that of non-BAX worms, indicating that PrP is able to antagonize BAX-mediated incapability of food sensing, likely through inhibiting BAX-induced cell destruction. To confirm that this anti-BAX effect is PrP specific, we generated a Pdat-1::PrP integrated line, cgIs58[Pdat-1(PrP)], in which PrP was only expressed in dopamine neurons. We crossed the lines of cgIs56[Pdat-1(BAX)] and cgIs58[Pdat-1(PrP)] and selected progeny co-expressing PrP and BAX in their dopamine neurons. As control, we also crossed cgIs56 with integrant lines of Pdat-1::GFP or Pdat-1::BI (cgIs59{Pdat-1[BAX inhibitor(BI)]}), respectively. As shown in , worms co-expressing Pdat-1::BAX and Pdat-1::PrP had a compatible food sensing ability with that of co-expressers of Pdat-1::BAX and Pdat-1::BI, demonstrating that PrP expressed in dopamine neurons was able to antagonize the BAX activity.
C. elegans has four bilaterally symmetric pairs of dopamine neurons, including two pairs of cephalic sensilla (CEP) neurons and one pair of anterior deirids (ADE) neurons in the head and one pair of posterior deirids (PDE) neurons in the posterior lateral position, all of which contain ciliated dendrites embedded in the cuticle and transmit mechanosensory information.Citation39 The simple structure and large size of C. elegans dopamine neurons make them easily visualized under a fluorescence microscope. As shown in , transgenic worms expressing Pdat-1::BAX exhibited dendrite discontinuities, large inclusions and small cell bodies in CEP and ADE neurons. However, co-expressers of Pdat-1::Bax/Pdat-1::PrP or Pdat-1::Bax/Pdat-1::BI showed healthy dopamine neurons with normal neuronal morphology and continuity, which were similar to that of GFP control worms (). Immunoblot analysis using specific antibodies confirmed the expression of BAX, PrP and BAX Inhibitor in corresponding integrated lines (). Taken together, our results indicate that PrP expression in dopamine neurons is able to inhibit BAX-induced cellular damage in live C. elegans, confirming that the mouse PrP has an anti-apoptotic function.
Anchorless PrP but not cytosolic PrP has an anti-BAX function.
Previously, it has been shown that transgenic mice expressing PrP mutant lacking the GPI-anchor were able to form infectious PrPSc amyloids but did not develop detectable neurological disorders. To test whether the GPI-anchorage is required for the anti-BAX function, we generated a Pdat-1::(gpi−) PrP integrated line (cgIs60[Pdat-1(gpi-PrP)]), in which the anchorless form of PrP was expressed (S3). The cgIs60 worms had a normal neuronal morphology and food sensing ability ( and data not shown). We crossed the lines of cgIs60 and cgIs56[Pdat-1(BAX)] followed by analyzing the food sensing ability of their progeny. As shown in , progeny co-expressing anchorless PrP and BAX in their dopamine neurons had normal food sensing ability, demonstrating that the GPI anchorage is not required for the anti-BAX function of PrP ().
A PrP truncation mutant, PrP(23–231) or cytoPrP, which lacks both signal sequence and GPI anchoring site, was shown to localize in the cytoplasm.Citation40 We next investigated whether cytoPrP is able to retain the anti-BAX function in C. elegans. We generated a Pdat-1::cytoPrP integrated line (cgIs61) expressing cytoPrP in dopamine neurons (S5). Although accumulation of cytoPrP in neuronal cell lines or transgenic mice led to cell death and prion-like neurological disordersCitation40 and expression of cyto-PrP in C. elegans muscles also resulted in mobility defect and paralysis,Citation26 we found that cytoPrP expression in C. elegans dopamine neurons did not affect their normal morphology and food sensing ability ( and data not shown). However, examining the food sensing ability of progeny obtained from the cross of cgIs61[Pdat-1(cytoPrP)] and cgIs56[Pdat-1(BAX)], we found that progeny co-expressing cytoPrP and BAX had lost their food sensing ability (), indicating that the cytosolic PrP failed to protect dopamine neurons from BAX-mediated cellular damage.
Discussion
The non-pathogenic nematode C. elegans presents several advantages as a promising model organism for prion study, including short lifecycle, simple nervous system, transparent body and genetic tractability. Previously, we showed that expression of the cytosolic form of mouse PrP, PrP(23–231), in the body wall muscles of C. elegans could result in abnormal body morphology, developmental defect and paralysis,Citation26 which are in agreement with early studies in mammals that cytoPrP expression is toxic.Citation40 In this study, we further expressed PrP in C. elegans neurons using the pan-neuronal promoter, Pric-19. Results from N-Glycosidase F and phosphatidylinositol-specific phospholipase C analyses indicated that PrP molecules produced in C. elegans neurons were properly glyclosylated and GPI-anchored ( and B). In situ immunofluorescence staining using a PrP monoclonal antibody also provided direct evidence that PrP molecules were localized on the cell surfaces of C. elegans neurons ( and Suppl. Movie 1). Our results, along with a very recent report by Bizat et al.Citation5 demonstrate that C. elegans can correctly recognize and execute the targeting instructions in a mammalian protein, suggesting that the cellular machinery responsible for PrP folding and processing is conserved between C. elegans and mammals.
Studies from wild-type PrP overexpression have provided important insights into prion toxicity. Overexpression of wild-type PrP in skeletal muscles caused sporadic inclusion-body myositis, dermatomyositis and neurogenic muscle atrophy, possibly due to accumulation of abnormal, cytotoxic forms of PrP.Citation35,Citation41,Citation42 In this study, we also showed that transgenic C. elegans with high PrP levels displayed abnormal morphology, striking neuropathogenic phenotypes and remarkable reductions in lifespan whereas C. elegans with low levels of PrP expression grew and developed normally ( and B). This finding is similar to a report by Chiesa et al. that transgenic mice with wild-type PrP overexpression had significantly shortened lifespan and developed spontaneous neurological disorders and synaptic abnormalities.Citation14 Although the underlying mechanisms for the observed lifespan reduction and other neuropathogenesis caused by PrP overexpression are not clear, results from Chiesa et al.Citation14 and this study seem to suggest that conformational differences of PrP might be the responsible mechanism. Mild resistance to proteinase K treatment and partial insolubility in 2% sarkosyl have been demonstrated for non-infectious, neurotoxic PrP aggregates resulted from wild-type PrP overexpression.Citation14 We also found that PrP in high expresser line cgIs51 showed a stronger proteinase K resistance than that in low expresser line cgIs55 (S4A). Moreover, the majority of PrP molecules in cgIs51 worms were in the pellet fraction in 2% sarkosyl after centrifugation whereas only a small percentage of PrP from cgIs55 worms was in the pellet fraction (S4B), suggesting that PrP molecules in the high expressers were less soluble, possibly, misfolded and aggregated. However, in situ thioflavin T (Th-T) staining of tissues prepared from both cgIs51 and cgIs55 worms showed no detectable amyloids (data not shown). Moreover, incubation of protein extracts from cgIs51 and cgIs55 with recombinant PrP monomers did not enhance PrP amyloid formation (data not shown). These results suggest that there were no PrP amyloid polymers formed in cgIs51 or cgIs55 worms. Rather, high expression likely causes PrP to misfold in C. elegans to result in formation of non-amyloid, neurotoxic oligomers that are mildly resistant to proteinase K and slightly insoluble in 2% sarkosyl.
Our phalloidin staining analysis shows that high expresser worms had normal muscle structures (S2), indicating that their mobility defects are possibly resulted from dysfunction of some unidentified motor neurons, which leads to obstruction of neuronal signal transmission to muscle cells. It is interesting to note that although the high expresser lines of cgIs51 and cgIs53 had similar levels of PrP expression, comparable PK-resistance and analogous solubility in 2% sarkosyl, cgIs53 worms exhibited more severe defects in mobility than cgIs51 ( and data not shown). While the mechanisms underneath the difference between cgIs51 and cgIs53 remain to be determined, we speculate that PrP molecules in these two lines might have adopted different toxic conformations to lead to the distinct phenotypes. The fact that both lines had similar expression levels of PrP also excludes the possibility that position effect—the Pric-19::PrP expression cassette was integrated into different chromosomal locations to result in different expression levels—is responsible for the observed phenotypic difference between cgIs51 and cgIs53.
A number of cellular functions have been implicated for the normal conformer of PrP, including copper binding,Citation43 iron uptake and transport,Citation44 membrane signaling,Citation45 neural cell adhesion,Citation46 synaptic transmissionCitation47 and protecting cells from oxidative stressCitation48 and apoptosis.Citation15 Taking advantage of the well-defined dopamine neuron system of C. elegans, we tested whether PrP expression can result in gain of anti-apoptotic function in transgenic PrP worms. Several key components of the apoptotic cascade have been identified in C. elegans, including the caspase CED-3, the APAF1 (apoptotic protease-activating factor-1) homolog CED-4 and the survival protein BCL-2 homologue CED-9.Citation49,Citation50 Systematic genetic analyses and cell culture studies have shown that these components interact to form a molecular framework crucial for apoptosis.Citation51,Citation52 In mammals, the pro-apoptotic factor BAX can undergo conformational activation leading to oligomerization and formation of a proteolipid pore in the outer mitochondrial membrane, which ultimately results in mitochondrial rupture and release of apoptogenic factors such as cytochrome c and activation of the caspase cascade.Citation53 Several studies suggest that CED-9 can have BCL-2 like (anti-apoptotic) or BAX-like (pro-apoptotic) activity, depending on its conformation.Citation54 In addition, loss of CED-9 activity inhibits mitochondrial fragmentation or cell death induced by the fission protein, dynamin-related protein 1 (DRP-1).Citation55 Although C. elegans does not contain a BAX ortholog, we have showed in this study that heterologous expression of mammalian BAX is able to induce cell damage in C. elegans dopamine neurons. It is unclear whether BAX stimulates the mitochondrial fission process through DRP-1, a key regulator of mitochondrial fission or competes with CED-4 against CED-9 to induce apoptosis in dopamine neurons. Our results from both behavioral and fluorescence microscopic assays show that BAX-induced dopamine neuron destruction in C. elegans can be rescued by mouse PrP coexpression (, C and ), confirming that PrP has an anti-BAX activity. Considering the fact that C. elegans does not have a PrP paralog, our clear-cut result supports the notion that PrPC is anti-apoptotic and it likely plays an important role in preventing neurodegeneration in mammals. Similar to two previous reports that ectopic expression of PrP lacking the GPI-anchorage prevented BAX-induced cell death in primary human neurons and yeast,Citation18,Citation56 we also found that a GPI-anchorless PrP mutant is able to rescue BAX-induced cell damage in C. elegans. Although immunoblot analysis indicates that the anchorless PrP and cytosolic PrP had similar expression levels (S3), cytosolic PrP failed to provide such protection in C. elegans (). Our result is contrary to what has been reported for human neurons.Citation57 This experimental discrepancy might be explained by that the cytoprotective function of PrP is not completely conserved from C. elegans to mammals. Another possibility might be that cytosolic PrP can sufficiently protect cultured neurons but not living animals from BAX-mediated destruction. Nevertheless, our finding that BAX is able to cause cellular damages in C. elegans and that PrP can function as a BAX inhibitor suggests that PrP can interact with important components in the apoptotic pathway in C. elegans. Given the versatility of C. elegans in genetic screens and manipulations, our C. elegans prion model can be valuable for investigating the potential role of PrP in apoptotic regulation since cellular components responsible for the cytoprotective activity of PrP should be readily identifiable.
Our integrated PrP transgenic lines may also be useful for testing other proposed biological functions of PrP. Since the C. elegans genome does not encode any PrP paralogs, PrP-mediated gain-of-functional pathways might be constituted in PrP-transgenics to allow developing simple, background-free functional assays. If successful, the well-defined neural system, transparent body and genetic tractability of C. elegans will likely facilitate the subsequent task of elucidating the responsible mechanisms underlying such PrP mediated gain-of-functions.
To understand how PrP misfolding leads to neurotoxicity remains the biggest challenge in prion research. It is intriguing that high expressions of wild-type PrP in mice, Drosophila and C. elegans all result in neurophathology and similar lifespan reduction (reviewed in ref. Citation14 and Citation22 and ). A transgenic Drosophila model with a high expression level of P101L, a mutant PrP that is linked to a familial prion disorder known as Gerstmann-Straussler-Scheinker syndrome, also showed locomotor dysfunction and lifespan reduction.Citation23 Such findings suggest that the three prion model organisms share a common cellular process from PrP misfolding to neurotoxicity and these prion models will likely prove valuable in identifying important components that are involved in this cellular process. In this regard, the molecular chaperone, Hsp70, has been shown to play an important role in preventing PrPSc-like conformer accumulation and subsequent neurodegeneration.Citation22 To understand the remarkable toxic phenotypes exhibited by our transgenic C. elegans expressing high levels of PrP will also likely provide insightful information on the responsible mechanisms for prion neuropathology and to facilitate the discovery of pharmacological compounds to suppress prion-mediated toxicity.
Materials and Methods
Strain and culture.
The Bristol N2 strain of C. elegans and its transgenic derivatives were cultured and maintained at 20°C as previously described.Citation26 All integrated strains were generated using the UV/TMP ultraviolet and trimethylpsoralen method.Citation27 Integrated C. elegans lines created in this study are described in .
Plasmids and constructs.
The coding sequence for mouse PrP(1-254) carrying the 3F4 epitope was PCR amplified from the PrP1-254-mPrP1,Citation28 using the following primers: 5′-GCG CGG CTA GCA TGG CGA ACC TTG GCT ACT GG-3′ (forward), 5′-GCG CGG AGC TCT CAT CCC ACG ATC AGG AAG ATG A-3′ (reverse). The resulting PCR products were digested with NheI/SacI and ligated to two plasmids that had been predigested with NheI/SacI: a modified pRIC19 (a gift from Dr. Ilya Ruvinsky, University of Chicago) in which the ATG codon in the first exon of ric19 is changed to AAG codon so that the original ATG in PrP open reading frame is used as the start codon for translation and pRB490, a plasmid containing the dopamine neuronal specific promoter Pdat-1,Citation29 to create Pric-19::PrP(1–254) and Pdat-1::PrP(1–254), respectively. To construct Pdat-1::BAX, pESC-LeuGAL10-mBax (a gift from Dr. David A. Harris, Boston University) was cleaved with NotI, filled with Klenow fragment and then digested with SacI. The resulting mBax fragment was then ligated to pRB490 that had been pretreated sequentially with NheI, Klenow and SacI. To generate the Pdat-1::mBax-inhibitor(mBI)-HA, p426GPD-mBI-HA (a gift from Dr. David A. Harris, Boston University) was digested with SpeI and SacI and the resulting mBI-HA fragment was cloned into the NheI and SacI sites of Pdat-1 plasmid. To construct Pdat-1::PrP(1–231) and Pdat-1::PrP(23–231), PCR fragments encoding anchorless PrP(1–231) or cytosolic PrP(23–231) were amplified with either 5′-GCG CGG CTA GCA TGG CGA ACC TTG GCT ACT GG-3′ (forward) and 5′-GCG CGG AGC TCC TAT TAG CTG GAT CTT CTC CCG-3′ (reverse) for anchorless PrP (1-231) or 5′-GCG CGG CTA GCA TGT CTA AAA AGC GGC CAA AGC CTG-3′ (forward) and 5′-GCG CGG AGC TCC TAT TAG CTG GAT CTT CTC CCG-3′ (reverse) for cytosolic PrP (23–231), using the PrP1-254-mPrP1 as the template. Both PCR products were digested with NheI/SacI and ligated into NheI/SacI predigested Pdat-1, respectively.
Protein electrophoresis and immunoblot analysis.
Animals collected from one large plate (15 cm) were frozen in liquid nitrogen and homogenized using a mini bead-beater (Biospec) in 300 µl of lysis buffer [made by mixing with 6 ml of 1× PBS, pH 7.0, 0.5% NP-40 and 1 tablet of protease inhibitor cocktail (Roche)]. Crude protein extracts were centrifuged at 11,000 rpm for 2 min at 4°C to remove the worm debris and unbroken worms. The supernatants were resolved by SDS-PAGE, immunoblotted with monoclonal PrP antibody (3F4) after transferred onto a PVDF membrane and detected with ECL kit (Amersham) as described previously.Citation26
Phalloidin staining and immunostaining.
The F-actin staining by Phalloidin and subsequent fluorescence detection were performed as described.Citation26 For PrP immunostaining, nematodes after freeze-crack treatmentCitation30 were first incubated with the 3F4 antibody followed by incubation of a TRITC-labeled secondary antibody. Images were collected with an Olympus (Tokyo, Japan) FV500 confocal microscope.
Behavioral assays.
Liquid thrashing assays were performed in 20 µl of M9 buffer as described.Citation26 Lifespan was measured according to a published protocol with minor modifications.Citation31 Briefly, embryos were synchronized by treatment of gravid adult worms with alkaline hypochlorite solution and allowed to develop to the L4 stage. L4 animals were then transferred to new plates and scored every other day. Animals were judged as dead by the lack of movement after prodding with a platinum wire. In all cases, at least 50 worms were used for each analysis. For pharyngeal pumping rates, the number of contractions of the terminal bulb of the pharynx was counted for 20 sec. Defecation cycle was the time between contractions of the posterior body-wall muscles and was measured for ten cycles. For each assay, ten animals were used for each genotype and their average values are shown.
Triton X-114 partitioning and phospholipase C treatment.
Two large plates (15 cm) of C. elegans were collected in M9 buffer. After centrifugation at 1,500 rpm for 2 min, the worm pellets were resuspended in 500 µl of lysis buffer. After addition of an equal volume of glass beads, the samples were cracked with liquid nitrogen three times, homogenized in a mini bead-beater (Biospec) and centrifuged at 14,000 rpm for 5 min at 4°C to remove the worm debris and unbroken worms. The supernatants were transferred to a clean tube. The Triton X-114 partitioning and phospholipase C treatment were performed as described.Citation21 Proteins in both aqueous and detergent phases were precipitated with methanol and subjected to SDS-PAGE and immunoblot analyses after resolubilization in 1× SDS sample buffer.
Proteinase K digestion and sarkosyl solubility assays.
Crude protein extracts were prepared from two large plates (15 cm) of worms expressing Pric-19::PrP in 300 µl of lysis buffer using a mini bead-beater as described before.Citation26 After centrifugation at 11,000× rpm for 2 min, the supernatant was digested with different concentrations (0, 1, 2, 5, 10 µg/ml) of Proteinase K at 37°C for 30 min before adding an equal volume of 2× SDS sample buffer to stop the reaction. For sarkosyl solubility assay, animals collected from two large plates (15 cm) were frozen in liquid nitrogen and homogenized using a mini bead-beater (Biospec) in 500 µl of lysis buffer. The sarkosyl solubility assays were performed as described before.Citation26 Protein samples from both Proteinase K digestion and sarkosyl solubility assays were examined by SDS-PAGE and immunoblot analysis.
Food-sensing behavior assay.
The locomotion rate was measured as described.Citation32 Well-fed, synchronized young adult animals (3 day old) were washed with M9 buffer. Animals were transferred to the center of an assay plate with or without bacterial lawn in a drop of M9 buffer. Five minutes after transfer, the rate of motion of each worm was counted in 20 sec intervals. For each assay, 15 animals were used for each genotype and their average values are shown.
RT-PCR.
Animals were removed from plates and washed two times with M9 buffer. Total RNA was extracted using RNeasy isolation kit (Qiagen). RNA concentrations were measured and equal amounts (500 ng) were used for reverse transcription. The qRT-PCR was performed according to the manufacturer's protocol (Invitrogen) with the appropriate forward and reverse gene-specific primers: 5′-CAT GAG CAG GCC CAT GAT CCA TTT-3′ (forward) and 5′-TGC ACG AAG TTG TTC TGG TTG CTG-3′ (reverse) for PrP or 5′-ACA GGT CAT CAC CGT TGG AAA CGA-3′ (forward) and 5′-ACG GTG TTG GCG TAC AAG TCC TTA-3′ (reverse) for ACT-4 as a control.
Figures and Tables
Figure 1 Creating transgenic C. elegans lines that express mouse prion protein (PrP). (A) Shown are Pric-19::GFP and Pric-19::PrP constructs, which contain the green fluorescence protein (green box) and the full-length PrP (1–254) (grey box) including the indicated N-terminal signal sequence (1–22) and the C-terminal site (231) for GPI anchoring, respectively. The ric-19 promoter (blue arrow) is also indicated. (B) After co-injection of the Pric-19::GFP and Pric-19::PrP constructs into the gonads of healthy young adult worms, five integrant lines expressing both GFP and PrP were obtained (see Materials and Methods). Shown are immunoblot analyses of the five lines using a PrP specific monoclonal antibody 3F4 and anti-actin. An integrant line expressing GFP alone was included as control.
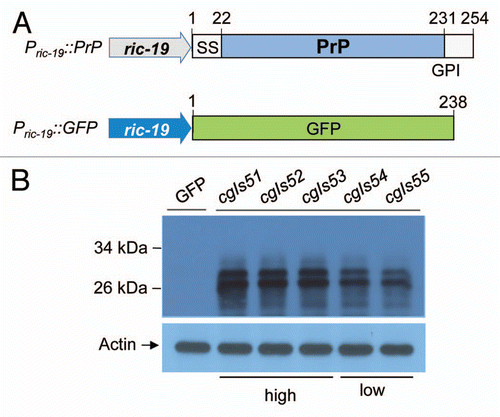
Figure 2 PrP molecules produced in C. elegans were glycosylated, GPI-anchored and presented on the plasma membranes of neurons. (A) Crude cell lysates prepared from high (cgIs51) and low (cgIs55) expressers were treated with (+) or without (−) PNGase F, which removes N-linked oligosaccharide chains from glycoproteins, followed by immunoblot analysis using the antibody 3F4. (B) Crude lysates from cgIs51 and cgIs55 were also treated with (+) or without (−) phosphatidylinositol-specific phospholipase C (PIPLC) for 2 h at 4°C, which releases GPI-anchored PrP molecules from the detergent phase, D, to the aqueous phase, A. After Triton X-114 treatment and phase partitioning, proteins were precipitated and immunoblotted with 3F4 antibody. (C) Nematodes co-expressing Pric-19::PrP and Pric-19::GFP were freeze-crack treated, incubated with the 3F4 antibody then a TRITC-labeled secondary antibody and followed by photographing under a fluorescence microscope. Left, PrP immunostaining; middle, ric-19::GFP expression; right, a merged image showing that GFP was localized in the cytoplasm whereas PrP was on the cell surface surrounding the GFP signal. Upper: high expresser line cgIs51; Lower: low expresser line cgIs55. The length of the bars in each image is 25 µm. The inset shows an enlarged motor neuron cell body on the ventral nerve cord (arrow heads).
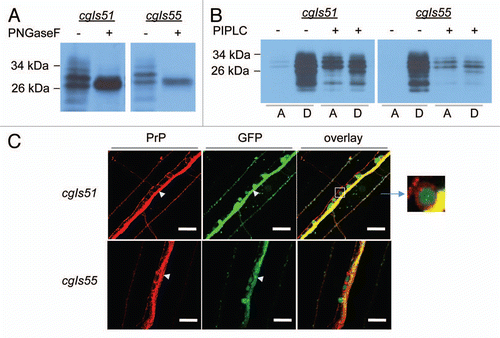
Figure 3 Morphologic and behavioral characteristics of transgenic worms expressing PrP in neurons. (A) Visualization of GFP expression in neurons of high (cgIs51 and cgIs53) and low (cgIs55) expressing lines of Pric-19::PrP/Pric-19::GFP. A transgenic worm expressing only Pric-19::GFP (GFP) was included as a control. The length of the bar in each image is 50 µm. As shown, the high expressers had dumpy-like body morphology: short and chunky-looking. (B) The high expressers (cgIs51 and cgIs53) also exhibited decreased lifespan whereas the low expressers (cgIs55) showed a lifespan comparable to that of non-transgenic control (N2) and worms expressing GFP only. Average lifespan in days (±SD) are: N2, 22.6 ± 2.5; GFP, 21.1 ± 1.8; cgIs51, 12.3 ± 1.8; cgIs53, 10.82 ± 2.5 and cgIs55, 19.3 ± 2.7. (C) Locomotion measured by liquid thrashing assays at adult stage for N2, GFP, cgIs51, cgIs53 and cgIs55worms. Each data point is the mean thrashing rate for ten worms. (D) Pharyngeal pumping rates were analyzed by counting terminal bulb contractions for a 20 sec period. (E) pBOC cycles were assayed on food by measuring the time between two posterior body contractions. For each assay, ten animals were used for each genotype and their average values are shown. *p < 0.01, in all cases, compared with the non-transgenic worms (N2) by Student's t-test.
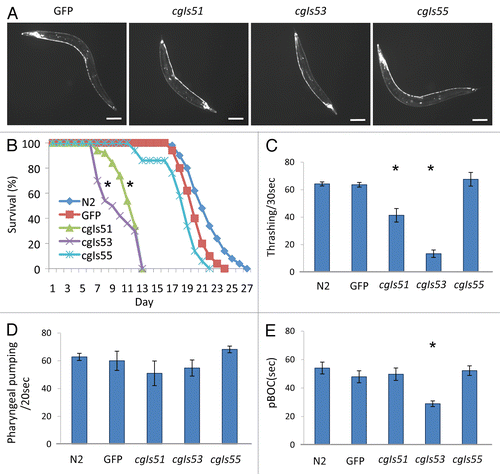
Figure 4 BAX expression in C. elegans dopamine neurons results in a comprised food sensibility and PrP inhibits BAX-mediated food sensibility reduction. (A) Food sensing behavioral assays of transgenic worms with their genotypes indicated, including two integrant lines of Pdat-1::BAX. The locomotion rates of 3-day-old worms were counted in the presence (with food) or absence (no food) of bacteria lawn. (B) Food sensing assay performed in worms coexpressing both PrP in whole neurons (Pric-19 based) and BAX in dopamine neurons (Pdat-1 based). (C) Food sensing assay in worms coexpressing PrP and BAX in dopamine neurons. *p < 0.01 by Student's t-test.
Figure 5 PrP inhibits BAX-induced cell destruction in C. elegans dopamine neurons. (A) Fluorescence microscopic examination of the dopamine neurons of living worms expressing Pdat-1::GFP. Worms with or without BAX, co-expressing PrP or BI-1 were examined and their CEP and ADE neurons are shown (arrows). All transgenes were expressed under Pdat-1. The length of the bar in each image is 25 µm. (B) Immunoblot analyses showing that PrP, BAX inhibitor and BAX were expressed in the integrant lines of cgIs56[dat-1(BAX)], cgIs58[dat-1(PrP)] and cgIs59[dat-1(BI)], respectively. (C) Food sensing assay carried out in worms co-expressing BAX and cytosolic PrP or BAX and anchorless PrP in dopamine neurons. *p < 0.01 by Student's t-test.
![Figure 5 PrP inhibits BAX-induced cell destruction in C. elegans dopamine neurons. (A) Fluorescence microscopic examination of the dopamine neurons of living worms expressing Pdat-1::GFP. Worms with or without BAX, co-expressing PrP or BI-1 were examined and their CEP and ADE neurons are shown (arrows). All transgenes were expressed under Pdat-1. The length of the bar in each image is 25 µm. (B) Immunoblot analyses showing that PrP, BAX inhibitor and BAX were expressed in the integrant lines of cgIs56[dat-1(BAX)], cgIs58[dat-1(PrP)] and cgIs59[dat-1(BI)], respectively. (C) Food sensing assay carried out in worms co-expressing BAX and cytosolic PrP or BAX and anchorless PrP in dopamine neurons. *p < 0.01 by Student's t-test.](/cms/asset/551693a7-c28a-496c-bfd6-c8a17c549826/kprn_a_10914026_f0005.gif)
Table 1 Integrant C. elegans strains used for this study
Additional material
Download Zip (1.4 MB)Acknowledgements
The authors thank I. Ruvensky for the gift of pRIC19 plasmid; D. Harris for the gifts of pESC-LeuGAL10-mBax and p426GPD-mBI-HA; J. Richmond and Anna Burdina for technical assistance; J. Kramer, J. Richmond and R. Nass for helpful discussions; members of the Li laboratory for critical comments; and E. Crow, R. Nass and C. Long for manuscript editing. This work was supported by grants from the United States Army (0850-370-R744), the Ellison Medical Foundation and the US National Institutes of Health (R01NS056086) to L.L.
References
- Aguzzi A, Heikenwalder M. Pathogenesis of prion diseases: current status and future outlook. Nat Rev Microbiol 2006; 4:765 - 775
- Prusiner SB. Novel proteinaceous infectious particles cause scrapie. Science 1982; 216:136 - 144
- Bendheim PE, Marmorstein AD, Potempska A, Bolton DC. Scrapie agent proteins do not accumulate in grey tremor mice. J Gen Virol 1988; 69:961 - 966
- Kretzschmar HA, Stowring LE, Westaway D, Stubblebine WH, Prusiner SB, Dearmond SJ. Molecular cloning of a human prion protein cDNA. DNA 1986; 5:315 - 324
- Manson J, West JD, Thomson V, McBride P, Kaufman MH, Hope J. The prion protein gene: a role in mouse embryogenesis?. Development 1992; 115:117 - 122
- Bueler H, Aguzzi A, Sailer A, Greiner RA, Autenried P, Aguet M, et al. Mice devoid of PrP are resistant to scrapie. Cell 1993; 73:1339 - 1347
- Legname G, Baskakov IV, Nguyen HO, Riesner D, Cohen FE, DeArmond SJ, et al. Synthetic mammalian prions. Science 2004; 305:673 - 676
- Wang F, Wang X, Yuan CG, Ma J. Generating a prion with bacterially expressed recombinant prion protein. Science 2010; 327:1132 - 1135
- Cobb NJ, Surewicz WK. Prion diseases and their biochemical mechanisms. Biochemistry 2009; 48:2574 - 2585
- Chesebro B, Trifilo M, Race R, Meade-White K, Teng C, LaCasse R, et al. Anchorless prion protein results in infectious amyloid disease without clinical scrapie. Science 2005; 308:1435 - 1439
- Chiesa R, Piccardo P, Quaglio E, Drisaldi B, Si-Hoe SL, Takao M, et al. Molecular distinction between pathogenic and infectious properties of the prion protein. J Virol 2003; 77:7611 - 7622
- Li A, Christensen HM, Stewart LR, Roth KA, Chiesa R, Harris DA. Neonatal lethality in transgenic mice expressing prion protein with a deletion of residues 105–125. EMBO J 2007; 26:548 - 558
- Solomon IH, Schepker JA, Harris DA. Prion neurotoxicity: Insights from prion protein mutants. Curr Issues Mol Biol 2009; 12:51 - 62
- Chiesa R, Piccardo P, Biasini E, Ghetti B, Harris DA. Aggregated, wild-type prion protein causes neurological dysfunction and synaptic abnormalities. J Neurosci 2008; 28:13258 - 13267
- Westergard L, Christensen HM, Harris DA. The cellular prion protein (PrP(C)): its physiological function and role in disease. Biochim Biophys Acta 2007; 1772:629 - 644
- Aguzzi A, Sigurdson C, Heikenwaelder M. Molecular mechanisms of prion pathogenesis. Annu Rev Pathol 2008; 3:11 - 40
- Kuwahara C, Takeuchi AM, Nishimura T, Haraguchi K, Kubosaki A, Matsumoto Y, et al. Prions prevent neuronal cell-line death. Nature 1999; 400:225 - 226
- Bounhar Y, Zhang Y, Goodyer CG, LeBlanc A. Prion protein protects human neurons against Bax-mediated apoptosis. J Biol Chem 2001; 276:39145 - 39149
- Flechsig E, Hegyi I, Leimeroth R, Zuniga A, Rossi D, Cozzio A, et al. Expression of truncated PrP targeted to Purkinje cells of PrP knockout mice causes Purkinje cell death and ataxia. EMBO J 2003; 22:3095 - 3101
- Bounhar Y, Mann KK, Roucou X, LeBlanc AC. Prion protein prevents Bax-mediated cell death in the absence of other Bcl-2 family members in Saccharomyces cerevisiae. FEMS Yeast Res 2006; 6:1204 - 1212
- Li A, Dong J, Harris DA. Cell surface expression of the prion protein in yeast does not alter copper utilization phenotypes. J Biol Chem 2004; 279:29469 - 29477
- Fernandez-Funez P, Casas-Tinto S, Zhang Y, Gomez-Velazquez M, Morales-Garza MA, Cepeda-Nieto AC, et al. In vivo generation of neurotoxic prion protein: role for hsp70 in accumulation of misfolded isoforms. PLoS Genet 2009; 5:e1000507
- Gavin BA, Dolph MJ, Deleault NR, Geoghegan JC, Khurana V, Feany MB, et al. Accelerated accumulation of misfolded prion protein and spongiform degeneration in a Drosophila model of Gerstmann-Straussler-Scheinker syndrome. J Neurosci 2006; 26:12408 - 12414
- Raeber AJ, Muramoto T, Kornberg TB, Prusiner SB. Expression and targeting of Syrian hamster prion protein induced by heat shock in transgenic Drosophila melanogaster. Mech Dev 1995; 51:317 - 327
- Bizat N, Peyrin JM, Haik S, Cochois V, Beaudry P, Laplanche JL, et al. Neuron dysfunction is induced by prion protein with an insertional mutation via a Fyn kinase and reversed by sirtuin activation in Caenorhabditis elegans. J Neurosci 2010; 30:5394 - 5403
- Park KW, Li L. Cytoplasmic expression of mouse prion protein causes severe toxicity in Caenorhabditis elegans. Biochem Biophys Res Commun 2008; 372:697 - 702
- Gengyo-Ando K, Mitani S. Characterization of mutations induced by ethyl methanesulfonate, UV and trimethylpsoralen in the nematode Caenorhabditis elegans. Biochem Biophys Res Commun 2000; 269:64 - 69
- Ma J, Lindquist S. De novo generation of a PrPSc-like conformation in living cells. Nat Cell Biol 1999; 1:358 - 361
- Nass R, Hall DH, Miller DM 3rd, Blakely RD. Neurotoxin-induced degeneration of dopamine neurons in Caenorhabditis elegans. Proc Natl Acad Sci USA 2002; 99:3264 - 3269
- Duerr JS, Frisby DL, Gaskin J, Duke A, Asermely K, Huddleston D, et al. The cat-1 gene of Caenorhabditis elegans encodes a vesicular monoamine transporter required for specific monoamine-dependent behaviors. J Neurosci 1999; 19:72 - 84
- Taub J, Lau JF, Ma C, Hahn JH, Hoque R, Rothblatt J, et al. A cytosolic catalase is needed to extend adult lifespan in C. elegans daf-C and clk-1 mutants. Nature 2003; 421:764
- Sawin ER, Ranganathan R, Horvitz HR. C. elegans locomotory rate is modulated by the environment through a dopaminergic pathway and by experience through a serotonergic pathway. Neuron 2000; 26:619 - 631
- Locht C, Chesebro B, Race R, Keith JM. Molecular cloning and complete sequence of prion protein cDNA from mouse brain infected with the scrapie agent. Proc Natl Acad Sci USA 1986; 83:6372 - 6376
- Stahl N, Borchelt DR, Hsiao K, Prusiner SB. Scrapie prion protein contains a phosphatidylinositol glycolipid. Cell 1987; 51:229 - 240
- Stimson E, Hope J, Chong A, Burlingame AL. Site-specific characterization of the N-linked glycans of murine prion protein by high-performance liquid chromatography/electrospray mass spectrometry and exoglycosidase digestions. Biochemistry 1999; 38:4885 - 4895
- Kraemer BC, Zhang B, Leverenz JB, Thomas JH, Trojanowski JQ, Schellenberg GD. Neurodegeneration and defective neurotransmission in a Caenorhabditis elegans model of tauopathy. Proc Natl Acad Sci USA 2003; 100:9980 - 9985
- Anderson P, Brenner S. A selection for myosin heavy chain mutants in the nematode Caenorhabditis elegans. Proc Natl Acad Sci USA 1984; 81:4470 - 4474
- Kindt KS, Quast KB, Giles AC, De S, Hendrey D, Nicastro I, et al. Dopamine mediates context-dependent modulation of sensory plasticity in C. elegans. Neuron 2007; 55:662 - 676
- Sulston J, Dew M, Brenner S. Dopaminergic neurons in the nematode Caenorhabditis elegans. J Comp Neurol 1975; 163:215 - 226
- Ma J, Wollmann R, Lindquist S. Neurotoxicity and neurodegeneration when PrP accumulates in the cytosol. Science 2002; 298:1781 - 1785
- Huang S, Liang J, Zheng M, Li X, Wang M, Wang P, et al. Inducible overexpression of wild-type prion protein in the muscles leads to a primary myopathy in transgenic mice. Proc Natl Acad Sci USA 2007; 104:6800 - 6805
- Sarkozi E, Askanas V, Engel WK. Abnormal accumulation of prion protein mRNA in muscle fibers of patients with sporadic inclusion-body myositis and hereditary inclusion-body myopathy. Am J Pathol 1994; 145:1280 - 1284
- Brown DR, Qin K, Herms JW, Madlung A, Manson J, Strome R, et al. The cellular prion protein binds copper in vivo. Nature 1997; 390:684 - 687
- Singh A, Kong Q, Luo X, Petersen RB, Meyerson H, Singh N. Prion protein (PrP) knock-out mice show altered iron metabolism: a functional role for PrP in iron uptake and transport. PLoS ONE 2009; 4:e6115
- Taylor DR, Hooper NM. The prion protein and lipid rafts. Mol Membr Biol 2006; 23:89 - 99
- Schmitt-Ulms G, Legname G, Baldwin MA, Ball HL, Bradon N, Bosque PJ, et al. Binding of neural cell adhesion molecules (N-CAMs) to the cellular prion protein. J Mol Biol 2001; 314:1209 - 1225
- Jeffrey M, McGovern G, Goodsir CM, Brown KL, Bruce ME. Sites of prion protein accumulation in scrapie-infected mouse spleen revealed by immuno-electron microscopy. J Pathol 2000; 191:323 - 332
- Milhavet O, Lehmann S. Oxidative stress and the prion protein in transmissible spongiform encephalopathies. Brain Res Brain Res Rev 2002; 38:328 - 339
- Ellis HM, Horvitz HR. Genetic control of programmed cell death in the nematode C. elegans. Cell 1986; 44:817 - 829
- Hengartner MO, Ellis RE, Horvitz HR. Caenorhabditis elegans gene ced-9 protects cells from programmed cell death. Nature 1992; 356:494 - 499
- Chinnaiyan AM, O'Rourke K, Lane BR, Dixit VM. Interaction of CED-4 with CED-3 and CED-9: a molecular framework for cell death. Science 1997; 275:1122 - 1126
- Hengartner MO. Programmed cell death in invertebrates. Curr Opin Genet Dev 1996; 6:34 - 38
- Lomonosova E, Chinnadurai G. BH3-only proteins in apoptosis and beyond: an overview. Oncogene 2008; 27:2 - 19
- Strasser A, O'Connor L, Dixit VM. Apoptosis signaling. Annu Rev Biochem 2000; 69:217 - 245
- Jagasia R, Grote P, Westermann B, Conradt B. DRP-1-mediated mitochondrial fragmentation during EGL-1-induced cell death in C. elegans. Nature 2005; 433:754 - 760
- Li A, Harris DA. Mammalian prion protein suppresses Bax-induced cell death in yeast. J Biol Chem 2005; 280:17430 - 17434
- Roucou X, Guo Q, Zhang Y, Goodyer CG, LeBlanc AC. Cytosolic prion protein is not toxic and protects against Bax-mediated cell death in human primary neurons. J Biol Chem 2003; 278:40877 - 40881