Abstract
By combining RNAi technology with SCNT method, we attempted to produce transgenic calves with knocked down bPRNP for technological assessments. The respective utilities of type II (tRNA) and type III (hU6) Pol III promoters in mediating plasmid vector-based RNAi for the production of a bPRNP-knockdown calf were compared. Plasmid harboring DNA for siRNA expression was introduced stably into the genome of primary cultured bovine cells. By inserting the transgenic cell into an enucleated bovine egg, SCNT embryos were produced. The ability for SCNT embryos to develop to blastocysts was higher in hU6 based vector groups (44–53%) than in a tRNA group (32%). In all, 30 hU6-embryos and 12 tRNA-embryos were transferred to 11 recipients. Only tRNA-embryos were able to impregnate recipients (6 out of 11 transfers), resulting in four aborted fetuses, one stillbirth, and one live-born calf. The expression of EGFP, a marker, was detected in all six. The bPRNP transcript levels in the nervous tissues (brain, cerebellum, spinal bulb, and spinal cord) from the calf, which was killed 20 days after birth, were reduced to 35% of those of the control calf on average, as determined by qRT-PCR. The PrPC levels, as estimated by western blot were reduced to 86% on average in the nervous tissues. These findings suggest that SCNT technology remains immature, that the tRNA promoter is useful, and that RNAi can significantly reduce PRNP mRNA levels, but insufficient reduction of PrPC levels exists in cattle under these conditions.
Introduction
Prion diseases, formally TSEs, are fatal neurodegenerative disorders that include CJD in humans, scrapie in sheep and BSE in cattle. These brain-degenerative diseases are caused by misfolding of PrPC into the protease-resistant isoform of PrPTSE. Conversion of PrPC to PrPTSE is believed to occur by interaction with exogenously introduced, self-replicating PrPTSE or by a very rare spontaneous event according to the protein-only hypothesis.Citation1
In fact, Prnp knockout mice are resistant to TSE, supporting this hypothesis.Citation2 It is particularly interesting that heterozygous Prnp(−/+) mice that express PrPC at about half of the normal level are practically resistant to TSE, as revealed by a significant delay in the onset and progression of clinical disease, although an increase in prion titer and PrPTSE levels was followed by symptoms of scrapie and death within weeks in wild-type animals.Citation3 These findings suggest that practically BSE-resistant cattle could be produced if PrPC expression were reduced by half or less. To achieve this, one method is to knockout bPRNP; the other is to knock down bPRNP using RNAi technology. In fact, bPRNP (−/−) knockout cattle have been produced using the sequential gene-targeting method.Citation4 Therefore, we attempted to knock down bPRNP using the RNAi technology to evaluate it.
To introduce an siRNA expression system into mammalian cells, plasmidsCitation4–Citation7 or lentivirus vectorsCitation8,Citation9 were used. Lentivirus vector-mediated RNAi reportedly suppresses prion protein in mice efficiently.Citation9–Citation11 At the start of this project, a P3 level facility, which is necessary to handle lentiviruses, was not available to us. We therefore used plasmid vectors, which can be handled routinely in a P1 or P2 level facility.
The siRNA systems using expression plasmids harboring an RNA polymerase III (Pol III) promoter, such as human small pronuclear promoter (hU6),Citation5,Citation6 human H1 promoter (H1),Citation12 or tRNA promoter,Citation13,Citation14 were shown to allow the expression of short hairpin RNA (shRNA) in mammalian cells. The Pol III promoters are classified into three different categories (Types I, II and III) based upon the promoter elements composition and their position relative to the transcriptional start site.Citation15 Type II promoters, such as tRNA promoter, have been used to express ribozymes and antisense RNA.Citation16,Citation17 Type III promoters, such as hU6 promoter, have a compact and simple organization; moreover, they are located upstream of transcribing regions. The siRNAs transcribed by the hU6 promoter reportedly suppress the expression of endogenous genes in several mammalian cell lines specifically.Citation5,Citation6 The choice of promoters is important for efficient RNAi in mammalian cells,Citation18,Citation19 but it is unclear which promoter is efficient in the in vivo expression of shRNA. Indeed, a U6 promoter was reported to be fatally hazardous in mouse experiments.Citation20 Therefore, we compared the utility of type II (tRNA) and type III (hU6) Pol III promoters in mediating plasmid vector-based RNAi in this study.
Results
Development of SCNT embryos in vitro.
For SCNT, stably transfected cells ( and B) were thawed and cultured for 3–4 days before transfer into bovine ova that had been matured in vitro. In many cases, EGFP expression was visualized immediately after nuclear transfer. However, the fluorescence was intense at the late embryonic stage and increased its intensity at the blastocyst stage (). Approximately 80% of couplets had fused and activated to become SCNT ova, whose cleavage ratios were, in decreasing order, ova with piGENE hU6, with piGENE hU6S/K and with piGENE tRNA (). Statistically significant differences were found in the ratios, but most SCNT ova showed cleavage, with the worst ratio being 86.8% (ova with piGENE tRNA). Cleaved ova were cultured for 4 days. The ability of the ova to develop to the blastocyst stage varied according to statistical significance, ranging from approximately 30–50%. The development ratios were, in decreasing order, embryos with piGENE hU6S/K, those with piGENE hU6, and those with piGENE tRNA (). Consequently, both cleavage and development ratios were higher in SCNT ova with the hU6 promoter vector groups than those with the tRNA group.
Development of SCNT blastocysts in vivo.
Only SCNT embryos with high quality (morphologically intact compacted morulae, small or no degree of fragmentation and/or unevenness in early and expanded blastocysts) 7 days after fusion were selected for embryo transfer. In all, 42 SCNT embryos were transferred into 11 recipients, yielding six positive gestations as determined by rectal palpation 60 days after embryo transfer (). Four fetuses were aborted at 120, 152, 156 and 185 days of gestation. All recipients with an aborted fetus displayed a moderate degree of hydroallantois. Two calves weighing 65 and 55 kg were born by cesarean section on day 287 and 292 of gestation, respectively, showing large offspring syndrome. Both calves were heavier than the average of females of this breed (38–45 kg). One of the two was a stillbirth; the live-born calf was unable to stand (). Because this calf had not been able to stand for 20 days, it was killed under deep anesthesia for additional analysis.
Analyses of transgenic fetus and calves.
Tissues of four aborted fetuses and two calves showed EGFP fluorescence under fluorescence microscopy (). In fact, EGFP fluorescence was detected in the brain (), cerebellum, spinal bulb, spinal cord, heart, lung, spleen and tongue of the live-born calf, indicating that EGFP driven by the CMV promoter was expressed in most or all tissues of the body. The EGFP gene was amplified using PCR from these four aborted fetuses and two calves, as expected ().
Sequencing analyses of PCR products showed that all four aborted fetuses and two calves harbored the piGENE tRNA construct (). The placenta and umbilical cord from the live-born calf also showed the presence of the piGENE tRNA construct. No mutation was found in their sequences. Regarding the proportional ratio of conceptions to the number of transferred embryos; 12 embryo transfers with the piGENE tRNA construct impregnated six recipients (50%), although none was impregnated using a total of 30 embryo transfers with the piGENE hU6 and piGENE hU6S/K construct. The difference in impregnation rates between tRNA and hU6 promoters is statistically significant (p = 0.0015).
bPRNP transcripts as measured using PCR.
When bPRNP transcript levels were estimated using qRT-PCR, several organs (brain, cerebellum, spinal bulb, spinal cord, lung and spleen) of the transgenic calf showed lessened levels of bPRNP mRNA (). The ratios of the relative levels of the transgenic calf to the control fluctuated from 0.080 (spleen) to 0.716 (spinal cord) depending on the organ. The mean ratios of four nervous tissues (brain, cerebellum, spinal bulb and spinal cord) were 0.347. Consequently, qRT-PCR revealed reduced mRNA levels of bPRNP in the transgenic calf. An end-point PCR experiment also revealed that the level of bPRNP mRNA in the brain of the transgenic calf was lower than that of the control (data not shown).
Western blot analysis for PrPC level.
The PrPC levels in several organs (brain, cerebellum, spinal bulb, spinal cord, lung and spleen) of the SCNT-clone and control calves were analyzed using western blot analysis. To estimate the PrPC levels, band intensities were compared with actin levels (). No tissues showed a statistically significant difference. The organs of the nervous system (brain, cerebellum, spinal bulb and spinal cord) showed reduced relative PrPC levels (85.6% on average), although other organs (spleen and lung) showed increased levels (139 and 159%, respectively).
PrPC levels in the brain of reference control cows.
bPRNP transcript and PrPC levels in the live-born transgenic calf were compared with those of a single age-, sex- and breed-matched calf. To estimate the levels in this control, the brains of six reference control cows that had been obtained from a farm were analyzed and used as reference controls for the live-born transgenic calf. Results are depicted in . The average PrPC levels of the six were 109% of the control. No statistically significant difference was found between the two, suggesting that the PrPC level of the control calf used here could be used as a relevant control, and that PrPC levels of the 20 day old control calf nearly reached those of adults.
Discussion
Actually, RNAi can mediate sequence-specific silencing of gene expression in widely various eukaryotes using siRNA including a sequence identical to the target gene.Citation21,Citation22 Along with concerns related to prion-mediated diseases in livestock and their potential transmission to humans, recent reports have described that RNAi can be exploited in silencing the expression of the prion protein in mice,Citation9–Citation11 a goat fetus and cattle blastocysts.Citation8 Lentivirus vectors have been report of its efficient. They were used in studies where shRNAs were driven by the Prnp promoter under the control of RNA pol IICitation9 or the H1 promoterCitation8,Citation10 or U6 promoterCitation11 under the control of RNA pol III. We used plasmid vectors harboring an shRNA expression system. Regarding impregnation ratios, plasmid vector systems are apparently inefficient; six conceptions were obtained from 42 embryo transfers (14%, ). Confined to embryos with the tRNA promoter, the ratio was 50% (6 of 12).
Promoter choices have been shown to be important for efficiency of RNAi because promoters direct high levels of shRNA transcription, which in turn is expected to mediate highly efficient silencing. In our previous study,Citation23 knocking down of bPRNP in cultured cells has shown that siRNA driven by the hU6 promoterCitation6,Citation7,Citation24 is slightly more effective than the tRNA promoter.Citation13,Citation14 Because no practical difference was apparent, we tried to determine which pol III promoter is better in siRNA-expressing embryo construction: hU6, modified hU6 (hU6S/K) or tRNA. Our present results demonstrated that the pol III type III promoter, hU6 and hU6S/K, has a significantly higher efficiency to develop SCNT embryos to blastocysts than the type II tRNA promoter (). To our surprise, only SCNT embryos with a tRNA-promoter construct impregnated recipients successfully, developed to term and produced a live-born calf ( and ), which, to our knowledge, is the first bPRNP-knockdown live-born calf.
The reason for the exclusive tRNA-promoter preference remains unknown. An intrinsic difference exists in nuclear exports of shRNA and tRNA. Ran-GTP and exportin 5 are coupled with shRNA and tRNA. Overexpression of shRNA saturates the activity of endogenous exportin 5.Citation25,Citation26 Indeed, the use of adeno-associated virus vectors expressing shRNA driven by hU6 or H1 promoter was toxic or fatal to mice,Citation20 suggesting the involvement of competitive use of exportin-5 by the shRNA and miRNA pathways. If this is the case in our experiment, then it is expected to occur during later developmental stages than the blastocyst stage. The U6 promoter was more potent than H1, and showed adverse effect on primary cells.Citation27 Consequently, potent promoters are not always preferred, underscoring the usefulness of the tRNA promoter in in vivo experiments.
A total of 42 transgenic blastocysts with an siRNA expression construct were transferred to 11 recipient heifers, resulting in six embryos impregnated. After day 120 of gestation, however, marked losses occurred: four fetuses were aborted with a hydroallantois symptom. Two full-term calves, still-birth and live-born, were overgrown and born by cesarean section. The live-born calf was unable to stand. Consequently, all showed anomalies to varied extents. In all fetuses and calves, EGFP was expressed intensively (). The EGFP expression, however, might not be the major cause of the anomalies because “green mice” are apparently normal.Citation28 In fact, large offspring syndrome (LOS) has been well known in domestic animals produced by assisted reproductive technology (ART).Citation29 In humans, nine imprinting syndromes have been identified: at least three are linked with ART.Citation30 Several changes in imprinting patterns in SCNT cattle have been reported for the SNRPN,Citation31 IGF-2R,Citation32 H19,Citation33 IGF2 and H19,Citation34 and PEG3 and MAOACitation35 genes. Failure in resetting of imprinted genes at SCNT must compose major factors affecting our aborted fetuses and abnormal calves. Technical innovations to reset imprinting conditions from somatic ones to embryonic ones are urgently needed.
Because PRNP/Prnp has been found in all animals examined (cattle, goats, hamsters, humans, mice, rats, sheep) as well as in chickens and even in yeasts,Citation36 it is expected to play some important roles. Involvement of PrPC in maintaining the brain's white matter and in regulating this tissue's innate immune cells, responses to oxidative stress and neuron formation is indicated.Citation37–Citation39 Recently, PrPC has been shown to be necessary to maintain the myelin sheath.Citation40 Several lines of evidence suggest that reducing prion protein expression by half could delay or even prevent disease progression,Citation3,Citation41,Citation42 because prion protein deficient mice are resistant to prion diseaseCitation43 and do not propagate infectious PrPTSE.Citation41 In this sense, the production of not knockout but knockdown calves with PrPBSE levels reduced sufficiently to prevent BSE progression is of interest. A qRT-PCR analysis showed that bPRPN mRNA levels were reduced to 8–72% of the control, depending on the organ (). The PrPC levels, however, were 79–93% of the control in the nervous tissues (). The roles of highly expressed PrPC in the spleen and lung are not known (). Turnover of PrPC might be higher in the nervous system than in other somatic organs. The overall difference in transcripts and protein levels suggests that the half life of PrPC is longer than that of mRNA. It is of interest to learn that PrPC levels in the brains of six cows were almost identical to those of the young control calf, implying the steady occurrence of PrPC, at least in the brain ().
Materials and Methods
siRNA expression vectors.
Construction of plasmid vectors for expression of siRNA to knocking down bPRNP was described previously in reference 23. The target sequence was GGG GAG AAC TTC ACC GAA ACT. The pEGFP-C1 vector (Clontech, Shiga, Japan) was modified by deleting the original MCS and inserting instead a new MCS consisting of Eco RI, Bgl II, Hind III and Bam HI at the Ase I site. Then, the promoter and the siRNA-coding DNA fragment of piGENE hU6, piGENE tRNA or piGENE hU6S/K constructCitation23 were introduced into the MCS. Plasmid DNA was purified, linearized and introduced into the genome of primary-cultured bovine lung cells.
Isolation and culture of bovine primary-cultured cells and transfection.
Primary cultured cells were obtained from the lung of a cross-bred fetus collected at a slaughterhouse. Cells were cultured in MEM α medium (α-MEM; Invitrogen Corp., Carlsbad, CA) supplemented with 10% (v/v) fetal bovine serum (FBS; Invitrogen Corp.,), 1% (v/v) MEM non-essential amino acid (Invitrogen Corp.,), and 50 µg/ml gentamicin (Sigma Chemical Co., St. Louis, MO). When the cells reached complete confluence, they were trypsinized and frozen for storage (in liquid nitrogen) or subcultured for transfection.
Cells (1.5 × 105) were plated in collagen type I-coated wells (six-well/plate; Asahi Techno Glass Corp., Funabashi, Japan) and transfected with 4 µg of linearized siRNA-expression pEGFP-C1 vector DNA. Transfection was performed using Lipofectamine 2000 (Invitrogen Corp.,) according to the manufacturer's instructions. Selection in 800 µg/ml of Geneticin (G418; Sigma) was initiated 24 h after transfection. After 7 days of selection, colonies were checked for EGFP expression using fluorescence microscopy (IX71; Olympus Corp., Tokyo). The EGFP-expressing cells were propagated and then frozen in a-MEM with 10% (v/v) dimethyl sulfoxide (DMSO; Wako Pure Chemical Industries Ltd., Osaka) until use.
Nuclear transfer.
After thawing, EGFP-expressing donor cells were cultured in a 4-well dish (Nunc A/S, Roskilde, Denmark) until 100% confluence, and maintained for 3–5 days prior SCNT manipulation. Immediately before transfer of a donor cell into enucleated oocytes, the monolayer of cells was washed twice with α-MEM without FBS and then incubated in 0.25% (w/v) trypsin-EDTA (Sigma) for 3 min at 38.5°C. After trypsinization, 3 ml of washing medium (α-MEM) supplemented with 10% FBS was added to neutralize trypsin activity. The cells were pelleted by centrifugation at 200 g for 5 min, re-suspended in the washing medium, and then kept until SCNT manipulation.
Preparation of recipient oocytes and SCNT was conducted according to the method described in reference Citation44. In brief, the bovine cumulus oocyte complexes (COCs) collected from the bovine ovaries were cultured for 19–22 h, and denuded in TCM 199 medium containing 0.1% (w/v) hyaluronidase (Sigma). The first polar body and adjacent nucleus visualized with Hoechst 33342 (3 µg/ml, Sigma) were removed from the oocyte using a beveled pipette. A transgenic donor cell was placed in the perivitelline space of the enucleated oocyte. The couplets were equilibrated for 3 min in the fusion buffer [0.3 M mannitol, 0.05 mM CaCl2 and 0.1 mM MgSO4, 0.5 mM HEPES and 0.4% (w/v) BSA], transferred into a drop of fusion buffer, and then manually aligned between the two electrode needles connected to the micromanipulator (MO-202D; Narishige, Tokyo). A single fusion and activation electrical pulse of 2.3 kV/cm for 30 µsec was applied to the couplets using Electro Cell Fusion Generator (LF101, NEPAGENE, Chiba, Japan). Successfully fused couplets were immediately activated in modified synthetic oviduct fluid (mSOF) medium,Citation45 supplemented with 0.4% (w/v) BSA and 10 µg/ml cycloheximide (Sigma), and incubated for 5 h. These were washed, transferred into mSOF medium supplemented with 0.4% (w/v) BSA, and then cultured for 72 h at 38.5°C in a portable incubator.Citation46 After 72 h of culture, only cleaved embryos were co-cultured with bovine cumulus cells in mSOF supplemented with 5% (v/v) FBS in 5% CO2 air at 38.5°C and cultured for an additional 4 days. The bovine cumulus cells had been prepared from recipient COCs. Percentages of embryos cleaved and developed to the blastocysts were recorded three and seven days after fusion, respectively. The blastocysts were visualized under a fluorescent microscope to determine whether they were transgenic as indicated by the expression of EGFP.
Synchronization of recipient heifers and embryo transfer.
Heat synchronization of the recipient heifers was induced by injecting 500 µg of prostaglandin F2α (Resipron-C, Teikokuzoki, Co., Ltd., Tokyo). After 6–7 days of standing heat, four to five fresh blastocysts were transferred into both uterine horns (two to three embryos per uterine horn). Because we compared the utility of type II (tRNA) and type III (hU6) Pol III promoters in mediating plasmid vector-based RNAi to clone transgenic cattle, embryos with different promoters (a total of 42 embryos; 12, 12 and 18 embryos with the piGENE tRNA and piGENE hU6 and piGENE hU6S/K constructs, respectively) were mixed in order to avoid biases due to recipients and transferred into the uterine horns of 11 heifers. Embryos were washed in mSOF supplemented with 5% (v/v) FBS, loaded into a 0.25 ml French plastic straw (two to three embryos per straw; I.M.V. L'Aigle, France) and then transferred to recipient heifers. The heifers were monitored daily for heat behavior and pregnancy was confirmed by rectal palpation 60 days after embryo transfer.
Transgene detection.
Detection of the transgene in cloned fetuses and calves was conducted using polymerase chain reaction (PCR). Briefly, commercially available bovine genomic DNA (Novagen Inc., WI) was used as the negative control. Tissue fragments from aborted fetuses, the stillbirth, the live-born calf, the dam and a control calf (25 days old) were collected. Then DNA was isolated using a DNeasy Tissue Kit (Qiagen Inc., MD) according to the manufacturer's instructions. The EGFP-specific primers used were: forward, 5′-TAT ATA GTC GAC CGT GTA CGG TGG GAG CTC TA-3′ and reverse, 5′-AAG CTT CTG CAG CTT GTA CAG CTC GTCVAT GC-3′. PCR primers for transgenes were: forward, 5′-TAT ATA GTC GAC TTT TTG TGA TGC TGA TGC TCG TCA GG-3′ and reverse, 5′-AAG CTT CTG CAG TTA TGT AAC GCG GAA CTC CA-3′. The Sal I site in the forward primer (underlined) and the Pst I site in the reverse (underlined) were used to insert the PCR product into the Bluescript SK(+) vector (Stratagene, CA) for sequencing. Template DNA was approximately 200 ng/tube. The DNA polymerase was KOD Dash (Toyobo Co., Ltd., Osaka). The PCR conditions were 94°C for 5 min, 35 cycles (1 min each) of denaturation at 95°C, annealing at 60°C and extension at 72°C and 72°C for 5 min. The sequencing apparatus was a model 3100 GeneScan (Applied Biosystems, CA).
Quantitative qRT-PCR.
The RNAs were isolated from the brain of the SCNT clone and control calves using a kit (RNeasy Protect Minikit; Qiagen Inc.) according to the manufacturer's instructions. Then cDNA was prepared from the RNA using a kit (PrimeScript RT Reagent Kit; Takara Bio Inc.). Power SYBR Green PCR Master Mix (Applied Biosystems) was used for qRT-PCR. The PCR primers were forward, 5′-CCA TGA CCA CTT TGG CAT C-3′ and reverse, 5′-GTT CAC GCC CAT CAC AAA C-3′ for GAPD (control) and forward, 5′-CTC TCG CAG AAG CAG GAC TT-3′ and reverse, 5′-ATG GCC ACA AAG AGA ACC AG-3′ for bPRNP. A mixture (5 µl of DNA, 5 µl of primer mix, 10 µl of reaction mix/well, triplicate reactions per sample) was applied to a sequence detection system (ABI Prism 7000; Applied Biosystems). The PCR conditions were 95°C for 10 min, 40 cycles of denaturation at 95°C for 10 s, annealing at 58°C for 50 s and synthesis at 72°C for 31 s. The standard curve was obtained by five dilutions of cDNA of the control brain from 0.1 to 0.00016 with a dilution coefficient of 5. Levels of GAPD and PRNP were calculated automatically from the standard curve. The relative ratios of PRNP cDNA to GAPD cDNA were obtained for tissues from the control and the transgenic calves. The relative values of PRNP/GAPD of the transgenic calf were compared with those of the control. Each datum was obtained from the mean of three reaction wells.
Western blot analysis.
The cloned calf was killed at 20 days old under deep anesthesia. Several tissues were dissected for analysis of bPRNP silencing by siRNA. Tissues of a 25 day old calf were also harvested for comparison. Approximately 200 mg of the samples were weighed and minced with five volumes of TBS buffer (Nacalai Tesque Inc., Kyoto). Samples were homogenized using a Polytron at 30,000 rpm for around 20 s on ice. An aliquot was used to determine protein concentrations by Bio-Rad Protein Assay (Bio-Rad Laboratories Inc., CA). Another aliquot (100 µl) was mixed with the same volume of Laemmli sample buffer (Bio-Rad Laboratories Inc.) containing 5% mercaptoethanol and boiled for 5 min. Samples were subjected to SDS-PAGE; then western blot analyses were conducted as described previously in reference Citation23. To quantify the intensity of each band, the integrated densities of PrPC and actin were measured using ImageJ (NIH). The relative intensity of PrPC was determined by dividing it with that of actin. The relative intensity of each organ of the transgenic calf was compared with that of the control animal.
Control animals.
A calf that was age-, sex- and breed-matched to the live-born transgenic calf was obtained from a farm and used as the control calf. To examine PrPC levels in the brain of the control calf, six brains of reference control cows were obtained from a slaughterhouse.
Statistical analyses.
The percentages of fused embryos, embryos cleaved and embryos developed to the blastocyst stage were analyzed using chi-square analysis. When some expected value was ≤0.05, Fisher's exact probability test was used. Differences at a probability p ≤ 0.05 were inferred as significant. Comparison of mean values was conducted using Student's t test.
Ethics.
The present animal experiments were approved by the Animal Research Committee of Yamaguchi University. Recombinant DNA experiments were conducted in accordance with regulations under the relevant laws of Japan.
Abbreviations
| ART | = | assisted reproductive technology |
| bPRNP | = | bovine prion gene |
| BSE | = | bovine spongiform encephalopathy |
| CJD | = | creutzfeldt-Jakob disease |
| CMV | = | cytomegalovirus |
| COCs | = | cumulus oocyte complexes |
| EGFP | = | enhanced green fluorescent protein |
| LOF | = | large offspring syndrome |
| Prnp | = | mouse prion gene |
| PrPBSE | = | bovine prion protein |
| PrPC | = | cellular prion protein |
| PrPTSE | = | prion protein of transmissible spongiform encephalopathy |
| qRT-PCR | = | quantitative reverse transcriptase polymerase chain reaction |
| RNAi | = | RNA interference |
| SCNT | = | somatic cell nuclear transfer |
| siRNA | = | short interfering RNA |
| TSE | = | transmissible spongiform encephalopathy |
Figures and Tables
Figure 1 Expression of EGFP in fetal lung cells transfected with siRNA expression plasmid vector DNA and in SCNT blastocysts. Lung cells (A and B), 8-cell stage (C and D) and blastocysts (E and F). Images of A, C and E were taken under visible light. Those of B, D and F were taken under fluorescent light. Bars represent 100 µm.
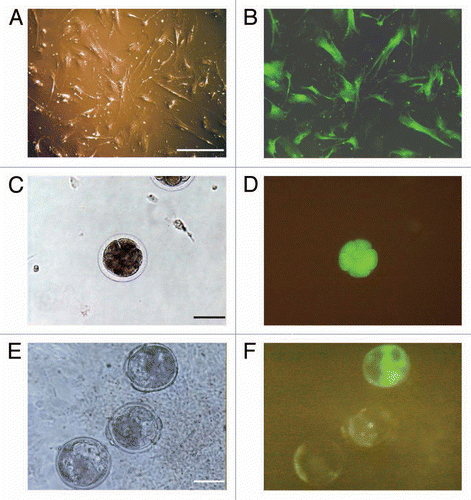
Figure 2 SCNT embryo-derived transgenic calf postpartum.

Figure 3 Expression of EGFP in transgenic fetuses and calves. A–G are phase contrast images taken under automatic exposure. H–N are fluorescent images of the same fields taken with a fixed exposure time of 1.8 s. A and H are the control calf (brain); aborted fetus No. 1 (muscle) (B and I); aborted fetus No. 2 (muscle) (C and J); aborted fetus No. 3 (muscle) (D and K); aborted fetus No. 4 (muscle) (E and L); the stillborn calf (muscle) (F and M) and live-born calf (brain) (G and N). All samples were stored under −80°C. A piece of a tissue was squashed between a slide glass and a glass slip and examined. The bar in (H) shows 100 µm.
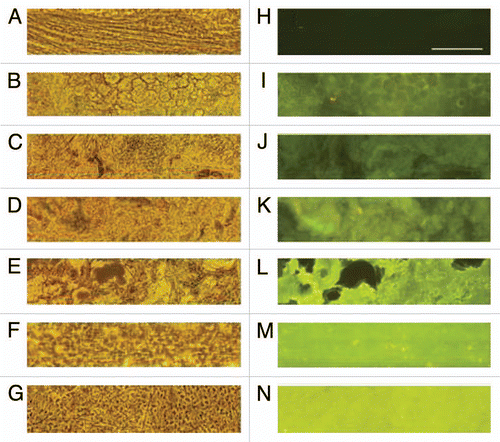
Figure 4 Relative bPRNP mRNA levels (PRNP/GAPD) in several organs of the transgenic calf as revealed by qRT-PCR. Gray columns show the control levels expressed as unity. Blank columns show those of the transgenic calf. Bars show the standard deviation of the mean. Asterisks denote a statistically significant difference from the control at less than a 5% (*) or 1% (**) level.
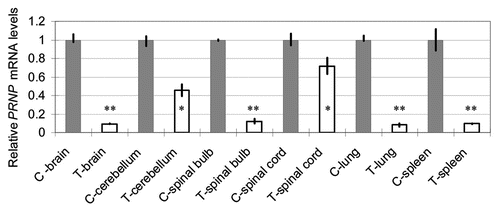
Figure 5 Relative PrPC levels (PrPC/actin) in several organs of the transgenic calf as revealed by western blotting. Gray columns show control levels expressed as unity. Blank columns show those of the transgenic calf. Bars show the range of three data obtained by application of 2.5, 5.0 and 10.0 µg proteins/lane. No statistically significant difference was found in any pair.
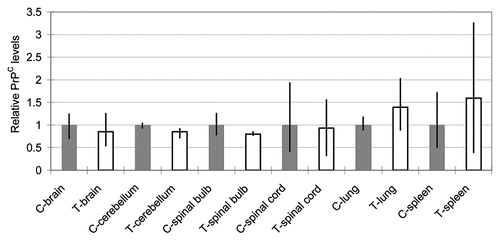
Figure 6 Relative PrPC levels (PrPC/actin) in the brain of reference cows as revealed by western blotting. The gray column shows the level of the control calf and blank columns, levels of reference cows. Bars show the average of two data obtained by application of 5.0 and 10.0 µg proteins/lane. No statistically significant difference was found between the control calf and reference cows.
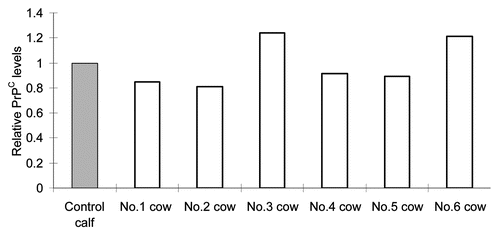
Table 1 Development of bovine embryos reconstructed with fetal lung cells transfected with siRNA expression plasmid vector DNA
Table 2 Overview of transgenic fetuses and calves with siRNA expression plasmid vector DNA
Acknowledgements
This study was mainly supported by a grant from Japan Science and Technology Agency to T.O. and in part by a grant from Shujitsu University to S.S.
References
- Prusiner SB. Molecular biology and transgenetics of prion diseases. Crit Rev Biochem Mol Biol 1991; 26:397 - 438
- Weissmann C. Molecular biology of transmissible spongiform encephalopathies. FEBS Lett 1996; 389:3 - 11
- Bueler H, Raeber A, Sailer A, Fischer M, Aguzzi A, Weissmann C. High prion and PrPSc levels but delayed onset of disease in scrapie-inoculated mice heterozygous for a disrupted PrP gene. Mol Med 1994; 1:19 - 30
- Richt JA, Kasinathan P, Hamir AN, Castilla J, Sathiyaseelan T, Vargas F, et al. Production of cattle lacking prion protein. Nat Biotechnol 2007; 25:132 - 138
- Lee NS, Dohjima T, Bauer G, Li H, Li MJ, Ehsani A, et al. Expression of small interfering RNAs targeted against HIV-1 rev transcripts in human cells. Nat Biotechnol 2002; 20:500 - 505
- Miyagishi M, Taira K. U6 promoter-driven siRNAs with four uridine 3′ overhangs efficiently suppress targeted gene expression in mammalian cells. Nat Biotechnol 2002; 20:497 - 500
- Paul CP, Good PD, Winer I, Engelke DR. Effective expression of small interfering RNA in human cells. Nat Biotechnol 2002; 20:505 - 508
- Golding MC, Long CR, Carmell MA, Hannon GJ, Westhusin ME. Suppression of prion protein in livestock by RNA interference. Proc Natl Acad Sci USA 2006; 103:5285 - 5290
- Gallozzi M, Chapuis J, Le Provost F, Le Dur A, Morgenthaler C, Peyre C, et al. Prnp knockdown in transgenic mice using RNA interference. Transgenic Res 2008; 17:783 - 791
- Pfeifer A, Eigenbrod S, Al-Khadra S, Hofmann A, Mitteregger G, Moser M, et al. Lentivector-mediated RNAi efficiently suppresses prion protein and prolongs survival of scrapie-infected mice. J Clin Invest 2006; 116:3204 - 3210
- White MD, Farmer M, Mirabile I, Brandner S, Collinge J, Mallucci GR. Single treatment with RNAi against prion protein rescues early neuronal dysfunction and prolongs survival in mice with prion disease. Proc Natl Acad Sci USA 2008; 105:10238 - 10243
- Hasuwa H, Kaseda K, Einarsdottir T, Okabe M. Small interfering RNA and gene silencing in transgenic mice and rats. FEBS Lett 2002; 532:227 - 230
- Hiraoka-Kanie M, Miyagishi M, Yamashita JK. Differentiation stage-specific analysis of gene function with inducible short hair-pin RNA in differentiating embryonic stem cells. Biochem Biophys Res Commun 2006; 351:669 - 674
- Kawasaki H, Taira K. Short hairpin type of dsRNAs that are controlled by tRNA(Val) promoter significantly induce RNAi-mediated gene silencing in the cytoplasm of human cells. Nucleic Acids Res 2003; 31:700 - 707
- Gunnery S, Ma Y, Mathews MB. Termination sequence requirements vary among genes transcribed by RNA polymerase III. J Mol Biol 1999; 286:745 - 757
- Sullenger BA, Lee TC, Smith CA, Ungers GE, Gilboa E. Expression of chimeric tRNA-driven antisense transcripts renders NIH 3T3 cells highly resistant to Moloney murine leukemia virus replication. Mol Cell Biol 1990; 10:6512 - 6523
- Yu M, Ojwang J, Yamada O, Hampel A, Rapapport J, Looney D, et al. A hairpin ribozyme inhibits expression of diverse strains of human immunodeficiency virus type 1. Proc Nat Acad Sci USA 1993; 90:6340 - 6344
- Boden D, Pusch O, Lee F, Tucker L, Shank PR, Ramratnam B. Promoter choice affects the potency of HIV-1 specific RNA interference. Nucleic Acids Res 2003; 31:5033 - 5038
- Makinen PI, Koponen JK, Karkkainen AM, Malm TM, Pulkkinen KH, Koistinaho J, et al. Stable RNA interference: comparison of U6 and H1 promoters in endothelial cells and in mouse brain. J Gene Med 2006; 8:433 - 441
- Grimm D, Streetz KL, Jopling CL, Storm TA, Pandey K, Davis CR, et al. Fatality in mice due to oversaturation of cellular microRNA/short hairpin RNA pathways. Nature 2006; 441:537 - 541
- Caplen NJ, Parrish S, Imani F, Fire A, Morgan RA. Specific inhibition of gene expression by small double-stranded RNAs in invertebrate and vertebrate systems. Proc Natl Acad Sci USA 2001; 98:9742 - 9747
- Elbashir SM, Harborth J, Lendeckel W, Yalcin A, Weber K, Tuschl T. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature 2001; 411:494 - 498
- Sutou S, Kunishi M, Kudo T, Wongsrikeao P, Miyagishi M, Otoi T. Knockdown of the bovine prion gene PRNP by RNA interference (RNAi) technology. BMC Biotechnol 2007; 7:44
- Brummelkamp TR, Bernards R, Agami R. A system for stable expression of short interfering RNAs in mammalian cells. Science 2002; 296:550 - 553
- Yi R, Doehle BP, Qin Y, Macara IG, Cullen BR. Overexpression of exportin 5 enhances RNA interference mediated by short hairpin RNAs and microRNAs. RNA 2005; 11:220 - 226
- Judge AD, Sood V, Shaw JR, Fang D, McClintock K, MacLachlan I. Sequence-dependent stimulation of the mammalian innate immune response by synthetic siRNA. Nat Biotechnol 2005; 23:457 - 462
- An DS, Qin FX, Auyeung VC, Mao SH, Kung SK, Baltimore D, et al. Optimization and functional effects of stable short hairpin RNA expression in primary human lymphocytes via lentiviral vectors. Mol Ther 2006; 14:494 - 504
- Ikawa M, Kominami K, Yoshimura Y, Tanaka K, Nishimune Y, Okabe M. A rapid and non-invasive selection of transgenic embryos before implantation using green fluorescent protein (GFP). FEBS Lett 1995; 375:125 - 128
- Young LE, Sinclair KD, Wilmut I. Large offspring syndrome in cattle and sheep. Rev Reprod 1998; 3:155 - 163
- Amor DJ, Halliday J. A review of known imprinting syndromes and their association with assisted reproduction technologies. Hum Reprod 2008; 23:2826 - 2834
- Suzuki J Jr, Therrien J, Filion F, Lefebvre R, Goff AK, Smith LC. In vitro culture and somatic cell nuclear transfer affect imprinting of SNRPN gene in pre- and post-implantation stages of development in cattle. BMC Dev Biol 2009; 9:9
- Long JE, Cai X. Igf-2r expression regulated by epigenetic modification and the locus of gene imprinting disrupted in cloned cattle. Gene 2007; 388:125 - 134
- Zhang S, Kubota C, Yang L, Zhang Y, Page R, O'Neill M, et al. Genomic imprinting of H19 in naturally reproduced and cloned cattle. Biol Reprod 2004; 71:1540 - 1544
- Curchoe CL, Zhang S, Yang L, Page R, Tian XC. Hypomethylation trends in the intergenic region of the imprinted IGF2 and H19 genes in cloned cattle. Anim Reprod Sci 2009; 116:213 - 225
- Liu JH, Yin S, Xiong B, Hou Y, Chen DY, Sun QY. Aberrant DNA methylation imprints in aborted bovine clones. Mol Reprod Dev 2008; 75:598 - 607
- Inoue Y. Life cycle of yeast prions: propagation mediated by amyloid fibrils. Protein Pept Lett 2009; 16:271 - 276
- Aguzzi A, Baumann F, Bremer J. The prion's elusive reason for being. Annu Rev Neurosci 2008; 31:439 - 477
- Sakudo A, Ikuta K. Fundamentals of prion diseases and their involvement in the loss of function of cellular prion protein. Protein Pept Lett 2009; 16:217 - 229
- Cobb NJ, Surewicz WK. Prion diseases and their biochemical mechanisms. Biochemistry 2009; 48:2574 - 2585
- Bremer J, Baumann F, Tiberi C, Wessig C, Fischer H, Schwarz P, et al. Axonal prion protein is required for peripheral myelin maintenance. Nat Neurosci 13:310 - 318
- Bueler H, Aguzzi A, Sailer A, Greiner RA, Autenried P, Aguet M, et al. Mice devoid of PrP are resistant to scrapie. Cell 1993; 73:1339 - 1347
- Mallucci G, Dickinson A, Linehan J, Klohn PC, Brandner S, Collinge J. Depleting neuronal PrP in prion infection prevents disease and reverses spongiosis. Science 2003; 302:871 - 874
- Bueler H, Fischer M, Lang Y, Bluethmann H, Lipp HP, DeArmond SJ, et al. Normal development and behaviour of mice lacking the neuronal cell-surface PrP protein. Nature 1992; 356:577 - 582
- Wongsrikeao P, Nagai T, Agung B, Taniguchi M, Kunishi M, Suto S, et al. Improvement of transgenic cloning efficiencies by culturing recipient oocytes and donor cells with antioxidant vitamins in cattle. Mol Reprod Dev 2007; 74:694 - 702
- Kwun J, Chang K, Lim J, Lee E, Lee B, Kang S, et al. Effects of exogenous hexoses on bovine in vitro fertilized and cloned embryo development: Improved blastocyst formation after glucose replacement with fructose in a serum-free culture medium. Mol Reprod Dev 2003; 65:167 - 174
- Suzuki T, Sumantri C, Khan NH, Murakami M, Saha S. Development of a simple, portable carbon dioxide incubator for in vitro production of bovine embryos. Anim Reprod Sci 1999; 54:149 - 157