Abstract
Amyotrophic lateral sclerosis (ALS) and frontotemporal lobar degeneration (FTLD) are clinically overlapping neurodegenerative disorders whose pathophysiology remains incompletely understood. ALS initiates in a discrete location, and typically progresses in a pattern consistent with spread of the degenerative process to involve neighboring regions of the motor system, although the basis of the apparent “spread” remains elusive. Recently mutations in two RNA binding proteins, TDP-43 and FUS, were identified in patients with familial ALS. In addition to being involved in numerous events related to RNA metabolism, each forms aggregates in neurons in ALS and FTLD. Recent evidence also indicates that both TDP-43 and FUS contain prion-related domains rich in glutamine (Q) and asparagine (N) residues, and in the case of TDP-43 this is the location of most disease causing mutations. This review discusses the potential relevance of the prion-related domains in TDP-43 and FUS in normal physiology, pathologic aggregation, and disease progression in ALS and FTLD.
First described in 1869, amyotrophic lateral sclerosis (ALS or Lou Gehrig disease) is one of the longest known neurodegenerative diseases.Citation1 The clinical presentation typically involves progressive weakness and muscle atrophy (due to degeneration of spinal motor neurons) and spasticity and reflex disinhibition (due to degeneration of upper motor neurons in the motor cortex) with death from respiratory failure within 3–5 years. Since the earliest descriptions by both Charcot and Gowers,Citation2 ALS progression was understood to have several key features. First is that it typically has a focal site of onset in the nervous system, i.e., begins with unilateral hand weakness. Second, progression is characterized by apparent “spread” of neurodegeneration, usually to the contralateral hand, followed by involvement of the legs. Recent detailed autopsy studies of ALS patients have confirmed that loss of motor neurons is most pronounced at the site of onset and diminishes in a gradient fashion with further distance from that site.Citation3 While many aberrant phenomena including excitotoxicity, oxidative stress, mitochondrial dysfunction and altered axonal transport have been implicated in ALS pathogenesis, it is not easily apparent how any of these could explain the focal initiation or the progressive spread of the disease through the motor system.Citation4
While the majority of ALS occurs sporadically, approximately 5–10% of patients have a family history of the disorder, typically autosomal dominant. For nearly 15 years the only known ALS gene was SOD1, mutations in which are responsible for ∼20% of familial cases. In 2006, accumulations of a RNA binding protein called TDP-43 were identified in degenerating neurons in both ALS and the clinically overlapping disorder fronto-temporal lobar degeneration (FTLD).Citation5 This was followed quickly by the identification of point mutations in TDP-43 in patients with familial ALS, indicating that altered TDP-43 function can be a primary cause of the disease.Citation6–Citation10 Shortly thereafter mutations in a second RNA binding protein called FUS were reported in familial ALS.Citation11,Citation12 Both TDP-43 and FUS are predominantly nuclear proteins involved in diverse aspects of RNA metabolism; however, in disease tissue both were observed to form inclusions in the cytosol of affected neurons. These findings suggested that aberrant protein aggregation may play a key role in ALS pathogenesis, akin to the central role of protein misfolding and aggregation observed in other neurodegenerative diseases. Interestingly, both FUS and TDP-43 contain “prion-related” Q/N rich domains and, in the case of TDP-43, essentially all of the ALS/FTLD associated mutations occur within this domain ().Citation13–Citation15 Although the importance of the prion-related domains in FUS and TDP-43 remains unclear, investigation into their role in the normal and pathologic functions of the proteins clearly warrants attention and is the focus of this review.
Prions and Prion-Related Domains
Prion protein remains the only known example of a protein capable of propagating a self-replicating conformation that can spread a disease (transmissible spongiform encephalopathy) across individuals and is thereby fitting of its name as an “infectious protein.”Citation16,Citation17 However, additional proteins exhibiting prion-like behavior are also observed in yeast, invertebrate and mammalian cells. In these cases the adoption of an alternate protein conformation and template based spreading of this conformation to the normal form, appears not to be deleterious and cause disease, but instead regulates the function of the aggregating protein.
Prion-like behavior of proteins is best characterized in yeast.Citation18,Citation19 The Sup35 protein is normally required for translational termination; however, under certain (particularly stressful) conditions it can form a self-propagating amyloid conformation transmissible to offspring, which is dependent on an intrinsically disordered region at the N-terminus particularly rich in glutamine (Q) and asparagine (N) residues.Citation20 Because this Q/N rich region is required for prion like propagation, it is referred to as the “prion domain.” Evidence supports that under stressful environmental conditions, induction of the Sup35 prion state leads to loss of Sup35 function and widespread read through of stop codons, allowing the rapid emergence of novel phenotypes.Citation21 Therefore, rather than representing a disease, prion domain mediated aggregation of Sup35 may actually be an adaptive strategy to provide immediate phenotypic diversity under stressful conditions.Citation19
The prion-domains of most yeast prions are similarly Q/N rich, including those in Ure2 and Rnq1, although others (including HET-s) are not. Therefore, while Q/N rich domains are permissive to allow a protein to adopt a prion-like conformational state, they are not absolutely required. A growing body of work has supported that while not all Q/N domain containing yeast proteins can function as prions, they share a strong tendency to self-aggregate when overexpressed.Citation22,Citation23
There is also evidence for prion like behavior of a Q/N rich protein in Aplysia.Citation24 CPEB is a RNA binding protein involved in regulating local synaptic protein synthesis. Synaptic activity appears to shift apCPEB from a monomeric to a multimeric form, which is dependent on the Q/N rich domain. In the multimeric form, apCPEB is active and regulates local mRNA translation to maintain synaptic facilitation. Similar behavior has also been observed in Drosophila where the prion-related Q/N domain of Pumilio, another RNA binding protein, regulates self-aggregation and post-synaptic translational suppression.Citation25
Finally, the mammalian genome contains a large number of proteins with Q/N rich prion related domains that may similarly use self-aggregation to modulate their activity.Citation26 A well studied example is the RNA-binding protein TIA-1, which is a key component of stress granules, cytoplasmic RNA-protein complexes formed under conditions of cellular stress which mediate mRNA translational suppression.Citation27 The prion related domain of TIA-1 is necessary for it to aggregate and organize stress granule formation.Citation28 A similar mechanism using Q/N domain mediated aggregation of RNA binding proteins also appears to be involved in the formation of P-bodies.Citation29
Therefore, a consistent theme for proteins containing prion-related Q/N rich domains from yeast through mammals is one of stimulus induced conformational change leading to self aggregation (often from environmental stress), which then alters protein function to organize an adaptive response (form stress granules, alter synaptic translation, etc.).
Evidence for Prion-Related Q/N Domains in TDP-43 and FUS
Given that prion-related domains are inherently disordered in structure, with minimal primary sequence determinants other than enrichment of Q/N residues, the presence of these domains was not immediately apparent in TDP-43 and FUS. TDP-43 structurally resembles heterogeneous nuclear ribonuclear proteins (hnRNPs), and it was originally noted to have a glycine-rich domain (residues 274–314) similar to other “RBD-Gly” family proteins, including hnRNPA1.Citation30,Citation31 Subsequently, some have referred to the entire C-terminal region as a glycine-rich domain analogous to hnRNPA1.Citation32 However, unlike hnRNPA1, the C-terminal domain of TDP-43 is not involved in binding to nucleic acids or nuclear shuttling.Citation31,Citation33,Citation34 Instead, the C-terminus of TDP-43 is required for it to function as a suppressor at several splicing targetsCitation35,Citation36 and for TDP-43 to act as a transcriptional insulator for the mouse sp-10 gene.Citation37 Importantly, all but one of the ALS associated mutations in TDP-43 occur in the C-terminal domain.
In an effort to define cellular stressors that regulate TDP-43 translocation from the nucleus, our group expressed several aggregation prone proteins in the cytosol of cultured cells, to determine if TDP-43 could act as a sensor for misfolded protein stress.Citation14 We observed that TDP-43 became tightly sequestered into detergent insoluble inclusions formed by polyglutamine proteins (Huntingtin N-terminal fragment or pure expanded polyglutamine), which required a particularly Q/N rich stretch of residues (31%) within the C-terminal domain of TDP-43. This region is similar in Q/N content to other proteins that were identified in unbiased screens for polyglutamine aggregate interacting proteins, including NF-Y, TIA-1 and FUS.Citation13,Citation38,Citation39 The presumed molecular basis of this interaction is the incorporation of the Q/N rich domain into the fibrillar β-sheet structure of the polyglutamine inclusion. This provided the first experimental evidence that the C-terminus of TDP-43 behaves similarly to other proteins with Q/N rich prion-related domains.
Inclusions of either TDP-43 or FUS are observed in cases of ALS and FTLD. For TDP-43, the C-terminal region is highly prone to aggregation, both as purified protein in vitroCitation40 or when expressed as a fragment in yeast or cultured mammalian cells.Citation41–Citation43 This strong tendency of the C-terminus of TDP-43 to self-associate and form aggregates is likewise consistent with the behavior of a prion-related Q/N domain containing protein.
Finally, several algorithms have been used to predict proteins that contain prion-related domains in both yeast and human genomes.Citation22,Citation23,Citation26 Most recently using a hidden Markov Model algorithm trained on known yeast prion domain containing proteins, FUS and TDP-43 were predicted as the fifteenth and sixty-ninth most likely to contain prion related domains out of nearly 30,000 proteins in the human genome.Citation15
Therefore there are currently three pieces of evidence that TDP-43 and FUS contain prion-related domains: (1) both have modular domains highly enriched in Q/N residues that meet prediction criteria for prion-related domains; (2) both have a strong tendency to self-associate and form aggregates and (3) both are effectively cross-seeded into polyglutamine inclusions, mediated by the Q/N rich region similar to other prion-related domain containing proteins like TIA-1.
Implications of Prion-Related Domains in the Normal Function of TDP-43 and FUS
As discussed above, a shared property of many proteins with prion-related Q/N domains is stimulus induced self aggregation, which alters the function of the protein in a switch like fashion. Therefore, a key question raised is whether TDP-43 and FUS undergo similar self aggregation in response to some type of stimulus and mediate a cellular response, akin to other Q/N rich domain containing proteins such as TIA-1. Both FUS and TDP-43 can be found in stress granules in cultured cells under certain conditions.Citation44–Citation49 However, neither TDP-43 nor FUS appear to be required for stress granule formation, and FUS is only present in these structures if it is mislocalized to the cytosol by mutations in the nuclear localization signal.Citation45,Citation47 Therefore it remains to be defined what role they actually play in stress granule mediated translational suppression. It is also important to point out that the same Q/N rich region of TDP-43 has been proposed to mediate protein-protein interactions with other components of the mRNA splicing machinery.Citation50 Therefore, the prion-related Q/N rich domain of TDP-43 may normally be involved in mediating protein-protein interactions for splicing regulation, and only in the disease state does it develop an alternate conformation that templates and seeds additional TDP-43 aggregation.
Implications of the Prion-Related Domains in TDP-43 and FUS in Neurodegeneration
It is important to consider that TDP-43 “pathology” (cytoplasmic TDP-43 inclusions, nuclear clearing) is not only observed in ALS and FTLD but is also frequently present in affected brain regions in Alzheimer disease, Parkinson disease, chronic traumatic encephalopathy and even inclusion body myopathies.Citation51–Citation54 This is quite consistent with the possibility that TDP-43 aggregation is part of a normal response to cellular stress, and is mediated by the prion-related domain. The TDP-43 inclusions themselves therefore may not be toxic or even protective, but instead are indicative of a stress response pathway involving TDP-43 that is activated in these cells. Therefore, defining a potential normal role of prion-domain mediated TDP-43 aggregation could help to explain the presence of TDP-43 aggregates in a wide variety of neurodegenerative conditions.
Another interesting implication of the presence of prion related Q/N domains in both TDP-43 and FUS is the known property of proteins with these domains to co-aggregate into inclusions of polyglutamine containing proteins.Citation13,Citation14 This observation provided the initial experimental evidence that TDP-43 contained a Q/N rich domain. Furthermore, sequestration of TDP-43 into polyglutamine inclusions led to a secondary loss of TDP-43 splicing regulation, and overexpression of TDP-43 rescued the toxicity of an N-terminal huntingtin fragment containing a polyglutamine expansion in cultured cells.Citation14 These findings are consistent with a model where TDP-43 and other Q/N rich proteins cross-seed with polyglutamine inclusions, which may be a mechanism of polyglutamine toxicity.Citation39 Recently, intermediate length polyglutamine expansions in Ataxin-2 were found to be associated with increased risk of ALS.Citation55 Although the details remain to be worked out, these findings further support the suggestion that there may be significant cross-talk between pathways of neurodegeneration in both polyglutamine diseases and ALS/FTLD, potentially mediated by the Q/N rich domains in TDP-43 and FUS.
Finally, the prion-related Q/N domains in TDP-43 and FUS have potential implications for the apparent “spread” of neurodegeneration throughout the motor system in ALS. Recently, increased attention has been focused on the concept that transmission of misfolded proteins involved in neurodegeneration (such as amyloid-β or tau) could be propagated from cell to cell in a prion-like fashion.Citation15,Citation56,Citation57 Although experimental evidence for this hypothesis is nascent at present, it is attractive as a potential explanation for the clinically observed spread of neurodegenerative diseases throughout particular neuronal networks. Given that prion-related Q/N domains are capable of developing altered conformers which recruit aggregation of the native protein, the presence of prion-related domains in TDP-43 and FUS provides a potential molecular substrate for transmission of aggregates of these proteins from cell to cell.
Conclusions
The discovery that TDP-43 and FUS play a key role in the pathogenesis of ALS and FTLD has been a significant breakthrough in understanding these diseases. It has emphasized the central role for protein misfolding, a common theme to most neurodegenerative diseases, and opened investigations into the role of altered RNA metabolism, which was previously unexplored. Although an intriguing finding, additional studies are clearly needed to delineate the importance of the prion-related Q/N domains in TDP-43 and FUS. These include determining what role they play in normal protein function, under what conditions they mediate pathologic aggregation of TDP-43 and FUS, and whether they might play a role in disease progression by allowing cell to cell transfer of pathologic protein aggregates.
Abbreviations
| ALS | = | amyotrophic lateral sclerosis |
| FTLD | = | frontotemporal lobar degeneration |
| TDP-43 | = | TAR DNA binding protein 43 kD |
| FUS | = | fused in sarcoma |
| RBD-Gly | = | RNA binding domain, glycine rich |
Figures and Tables
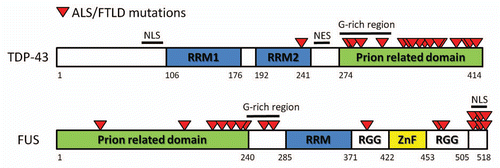
Figure 1 Line diagrams of TDP-43 and FUS showing the relationship between the prion-related domains and mutations in ALS and FTLD. The location of the prion-related domains are based on experimental findings of their interactions with polyglutamine inclusionsCitation13,Citation14 and a prediction algorithm based on yeast prion domains.Citation15 In the case of TDP-43, all but one of the ALS associated mutations are located in the prion-related Q/N rich domain. In FUS, the majority of ALS associated mutations occur in the C-terminal nuclear localization signal (NLS). However, a second cluster also occurs in or adjacent to the N-terminal prion related domain. NES, nuclear export signal; RRM, RNA binding domain; RGG, arginine, glycine, glycine repeat rich region; ZnF, zinc finger domain.
Acknowledgements
R.H.B. is supported by the NIH/NINDS (K08 NS055980 and R01 NS069669), the Muscular Dystrophy Association (135428), the Children's Discovery Institute and holds a Career Award for Medical Scientists from the Burroughs Wellcome Fund.
References
- Charcot JM, Joffroy A. Deux cas d'atrophie musculaire progressive avec les lesions de la substance grise et faiscaux anterolateraux de la moelle epiniere. Archives de Physiologic Normale et Pathologique 1869; 2:354 - 367
- Gowers WR. A manual of diseases of the nervous system 1886; London Churchill
- Ravits J, Laurie P, Fan Y, Moore DH. Implications of ALS focality: rostral-caudal distribution of lower motor neuron loss postmortem. Neurology 2007; 68:1576 - 1582
- Ravits JM, La Spada AR. ALS motor phenotype heterogeneity, focality and spread: deconstructing motor neuron degeneration. Neurology 2009; 73:805 - 811
- Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2006; 314:130 - 133
- Gitcho MA, Baloh RH, Chakraverty S, Mayo K, Norton JB, Levitch D, et al. TDP-43 A315T mutation in familial motor neuron disease. Ann Neurol 2008; 63:535 - 538
- Yokoseki A, Shiga A, Tan CF, Tagawa A, Kaneko H, Koyama A, et al. TDP-43 mutation in familial amyotrophic lateral sclerosis. Ann Neurol 2008; 63:538 - 542
- Sreedharan J, Blair IP, Tripathi VB, Hu X, Vance C, Rogelj B, et al. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 2008; 319:1668 - 1672
- Van Deerlin VM, Leverenz JB, Bekris LM, Bird TD, Yuan W, Elman LB, et al. TARDBP mutations in amyotrophic lateral sclerosis with TDP-43 neuropathology: a genetic and histopathological analysis. Lancet Neurol 2008; 7:409 - 416
- Kabashi E, Valdmanis PN, Dion P, Spiegelman D, McConkey BJ, Vande Velde C, et al. TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat Genet 2008; 40:572 - 574
- Kwiatkowski TJ Jr, Bosco DA, Leclerc AL, Tamrazian E, Vanderburg CR, Russ C, et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 2009; 323:1205 - 1208
- Vance C, Rogelj B, Hortobagyi T, De Vos KJ, Nishimura AL, Sreedharan J, et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 2009; 323:1208 - 1211
- Doi H, Okamura K, Bauer PO, Furukawa Y, Shimizu H, Kurosawa M, et al. RNA-binding protein TLS is a major nuclear aggregate-interacting protein in huntingtin exon 1 with expanded polyglutamine-expressing cells. J Biol Chem 2008; 283:6489 - 6500
- Fuentealba RA, Udan M, Bell S, Wegorzewska I, Shao J, Diamond MI, et al. Interaction with polyglutamine aggregates reveals a Q/N rich domain in TDP-43. J Biol Chem 2010; 285:26304 - 26314
- Cushman M, Johnson BS, King OD, Gitler AD, Shorter J. Prion-like disorders: blurring the divide between transmissibility and infectivity. J Cell Sci 2010; 123:1191 - 1201
- Griffith JS. Self-replication and scrapie. Nature 1967; 215:1043 - 1044
- Prusiner SB. Novel proteinaceous infectious particles cause scrapie. Science 1982; 216:136 - 144
- Wickner RB, Edskes HK, Roberts BT, Baxa U, Pierce MM, Ross ED, et al. Prions: proteins as genes and infectious entities. Genes Dev 2004; 18:470 - 485
- Halfmann R, Alberti S, Lindquist S. Prions, protein homeostasis and phenotypic diversity. Trends Cell Biol 2010; 20:125 - 133
- Wickner RB, Shewmaker F, Kryndushkin D, Edskes HK. Protein inheritance (prions) based on parallel in-register beta-sheet amyloid structures. Bioessays 2008; 30:955 - 964
- Tyedmers J, Madariaga ML, Lindquist S. Prion switching in response to environmental stress. PLoS Biol 2008; 6:294
- Michelitsch MD, Weissman JS. A census of glutamine/asparagine-rich regions: implications for their conserved function and the prediction of novel prions. Proc Natl Acad Sci USA 2000; 97:11910 - 11915
- Alberti S, Halfmann R, King O, Kapila A, Lindquist S. A systematic survey identifies prions and illuminates sequence features of prionogenic proteins. Cell 2009; 137:146 - 158
- Si K, Lindquist S, Kandel ER. A neuronal isoform of the aplysia CPEB has prion-like properties. Cell 2003; 115:879 - 891
- Salazar AM, Silverman EJ, Menon KP, Zinn K. Regulation of synaptic Pumilio function by an aggregation-prone domain. J Neurosci 2010; 30:515 - 522
- Harrison PM, Gerstein M. A method to assess compositional bias in biological sequences and its application to prion-like glutamine/asparagine-rich domains in eukaryotic proteomes. Genome Biol 2003; 4:40
- Anderson P, Kedersha N. RNA granules: post-transcriptional and epigenetic modulators of gene expression. Nat Rev Mol Cell Biol 2009; 10:430 - 436
- Gilks N, Kedersha N, Ayodele M, Shen L, Stoecklin G, Dember LM, et al. Stress granule assembly is mediated by prion-like aggregation of TIA-1. Mol Biol Cell 2004; 15:5383 - 5398
- Reijns MA, Alexander RD, Spiller MP, Beggs JD. A role for Q/N-rich aggregation-prone regions in P-body localization. J Cell Sci 2008; 121:2463 - 2472
- He Y, Smith R. Nuclear functions of heterogeneous nuclear ribonucleoproteins A/B. Cell Mol Life Sci 2009; 66:1239 - 1256
- Ou SH, Wu F, Harrich D, Garcia-Martinez LF, Gaynor RB. Cloning and characterization of a novel cellular protein, TDP-43, that binds to human immunodeficiency virus type 1 TAR DNA sequence motifs. J Virol 1995; 69:3584 - 3596
- Pesiridis GS, Lee VM, Trojanowski JQ. Mutations in TDP-43 link glycine-rich domain functions to amyotrophic lateral sclerosis. Hum Mol Genet 2009; 18:156 - 162
- Buratti E, Baralle FE. Characterization and functional implications of the RNA binding properties of nuclear factor TDP-43, a novel splicing regulator of CFTR exon 9. J Biol Chem 2001; 276:36337 - 36343
- Winton MJ, Igaz LM, Wong MM, Kwong LK, Trojanowski JQ, Lee VM. Disturbance of nuclear and cytoplasmic Tar DNA binding protein (TDP-43) induces disease-like redistribution, sequestration and aggregate formation. J Biol Chem 2008; 19:13302 - 13309
- Buratti E, Brindisi A, Giombi M, Tisminetzky S, Ayala YM, Baralle FE. TDP-43 binds heterogeneous nuclear ribonucleoprotein A/B through its C-terminal tail: an important region for the inhibition of cystic fibrosis transmembrane conductance regulator exon 9 splicing. J Biol Chem 2005; 280:37572 - 37584
- Ayala YM, Pantano S, D'Ambrogio A, Buratti E, Brindisi A, Marchetti C, et al. Human, Drosophila and C. elegans TDP43: nucleic acid binding properties and splicing regulatory function. J Mol Biol 2005; 348:575 - 588
- Abhyankar MM, Urekar C, Reddi PP. A novel CpG-free vertebrate insulator silences the testis-specific SP-10 gene in somatic tissues: role for TDP-43 in insulator function. J Biol Chem 2007; 282:36143 - 36154
- Yamanaka T, Miyazaki H, Oyama F, Kurosawa M, Washizu C, Doi H, et al. Mutant Huntingtin reduces HSP70 expression through the sequestration of NF-Y transcription factor. EMBO J 2008; 27:827 - 839
- Furukawa Y, Kaneko K, Matsumoto G, Kurosawa M, Nukina N. Cross-seeding fibrillation of Q/N-rich proteins offers new pathomechanism of polyglutamine diseases. J Neurosci 2009; 29:5153 - 5162
- Johnson BS, Snead D, Lee JJ, McCaffery JM, Shorter J, Gitler AD. TDP-43 is intrinsically aggregation-prone and amyotrophic lateral sclerosis-linked mutations accelerate aggregation and increase toxicity. J Biol Chem 2009; 284:20329 - 20339
- Johnson BS, McCaffery JM, Lindquist S, Gitler AD. A yeast TDP-43 proteinopathy model: Exploring the molecular determinants of TDP-43 aggregation and cellular toxicity. Proc Natl Acad Sci USA 2008; 105:6439 - 6444
- Igaz LM, Kwong LK, Chen-Plotkin A, Winton MJ, Unger TL, Xu Y, et al. Expression of TDP-43 C-terminal fragments in vitro recapitulates pathological features of TDP-43 proteinopathies. J Biol Chem 2009; 284:8516 - 8524
- Zhang YJ, Xu YF, Cook C, Gendron TF, Roettges P, Link CD, et al. Aberrant cleavage of TDP-43 enhances aggregation and cellular toxicity. Proc Natl Acad Sci USA 2009; 106:7607 - 7612
- Andersson MK, Stahlberg A, Arvidsson Y, Olofsson A, Semb H, Stenman G, et al. The multifunctional FUS, EWS and TAF15 proto-oncoproteins show cell type-specific expression patterns and involvement in cell spreading and stress response. BMC Cell Biol 2008; 9:37
- Colombrita C, Zennaro E, Fallini C, Weber M, Sommacal A, Buratti E, et al. TDP-43 is recruited to stress granules in conditions of oxidative insult. J Neurochem 2009; 111:1051 - 1061
- Freibaum BD, High AA, Chitta R, Taylor JP. Global analysis of TDP-43 interacting proteins reveals strong association with RNA splicing and translation machinery. J Proteome Res 2010; 9:1104 - 1120
- Dormann D, Rodde R, Edbauer D, Bentmann E, Fischer I, Hruscha A, et al. ALS-associated fused in sarcoma (FUS) mutations disrupt Transportin-mediated nuclear import. EMBO J 2010; 29:2841 - 2857
- Gal J, Zhang J, Kwinter DM, Zhai J, Jia H, Jia J, et al. Nuclear localization sequence of FUS and induction of stress granules by ALS mutants. Neurobiol Aging 2010; In press
- Bosco DA, Lemay N, Ko HK, Zhou H, Burke C, Kwiatkowski TJ Jr, et al. Mutant FUS proteins that cause amyotrophic lateral sclerosis incorporate into stress granules. Hum Mol Genet 2010; 19:4160 - 4175
- D'Ambrogio A, Buratti E, Stuani C, Guarnaccia C, Romano M, Ayala YM, et al. Functional mapping of the interaction between TDP-43 and hnRNP A2 in vivo. Nucleic Acids Res 2009; 37:4116 - 4126
- Josephs KA, Whitwell JL, Knopman DS, Hu WT, Stroh DA, Baker M, et al. Abnormal TDP-43 immunoreactivity in AD modifies clinicopathologic and radiologic phenotype. Neurology 2008; 70:1850 - 1857
- Nakashima-Yasuda H, Uryu K, Robinson J, Xie SX, Hurtig H, Duda JE, et al. Co-morbidity of TDP-43 proteinopathy in Lewy body related diseases. Acta Neuropathol 2007; 114:221 - 229
- King A, Sweeney F, Bodi I, Troakes C, Maekawa S, Al-Sarraj S. Abnormal TDP-43 expression is identified in the neocortex in cases of dementia pugilistica, but is mainly confined to the limbic system when identified in high and moderate stages of Alzheimer's disease. Neuropathology 2010; 30:408 - 419
- Weihl CC, Temiz P, Miller SE, Watts G, Smith C, Forman M, et al. TDP-43 accumulation in inclusion body myopathy muscle suggests a common pathogenic mechanism with frontotemporal dementia. J Neurol Neurosurg Psychiatry 2008; 79:1186 - 1189
- Elden AC, Kim HJ, Hart MP, Chen-Plotkin AS, Johnson BS, Fang X, et al. Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALS. Nature 2010; 466:1069 - 1075
- Frost B, Diamond MI. Prion-like mechanisms in neurodegenerative diseases. Nat Rev Neurosci 2010; 11:155 - 159
- Brundin P, Melki R, Kopito R. Prion-like transmission of protein aggregates in neurodegenerative diseases. Nat Rev Mol Cell Biol 2010; 11:301 - 307