Abstract
Neurodegenerative diseases are caused by proteinaceous aggregates, usually consisting of misfolded proteins which are often typified by a high proportion of β-sheets, which accumulate in the Central Nervous System. These diseases, including Morbus Alzheimer, Parkinson disease and Transmissible Spongiform Encephalopathies (TSEs) also termed prion disorders, afflict a substantial proportion of the human population and as such the etiology and pathogenesis of these diseases has been the focus of mounting research. Although many of these diseases arise from genetic mutations or are sporadic in nature, the possible horizontal transmissibility of neurodegenerative diseases poses a great threat to population health. In this article we discuss recent studies which suggest that the “non-transmissible” status bestowed upon Alzheimer and Parkinson diseases may need to be revised as these diseases have been successfully induced through tissue transplants. Furthermore, we highlight the importance of investigating the “natural” mechanism of prion transmission including peroral and perenteral transmission, proposed routes of gastrointestinal uptake and neuroinvasion of ingested infectious prion proteins. We examine the multitude of factors which may influence oral transmissibility and discuss the zoonotic threats which Chronic Wasting Disease (CWD), Bovine Spongiform Encephalopathy (BSE) and Scrapie may pose resulting in vCJD or related disorders. In addition, we suggest that the 37 kDa/67 kDa laminin receptor on the cell surface of enterocytes, a major cell population in the intestine, may play an important role in the intestinal pathophysiology of alimentary prion infections.
Many different mechanisms exist which underlie the etiology of the numerous neurodegenerative diseases affecting the human population. Amongst the most prominent are Morbus Alzheimer, prion disorders, Parkinson disease, Chorea Huntington, frontotemporal dementia and amylotrophic lateral sclerosis. The molecular mechanisms underlying these diseases vary; however, all neurodegenerative diseases share a common feature: they are caused by protein aggregation. The only neurodegenerative diseases proven to be transmissible are prion disorders. In contrast to frontotemporal dementia, recent evidence suggests that Alzheimer and Parkinson diseases may also be transmissible. Pre-symptomatic Alzheimer disease (APP23) mice exhibited an increase in the Alzheimer phenotype when brain homogenate of autopsied human Alzheimer disease patients and older, amyloid beta- (Aβ-) laden APP23 mice was injected into their hippocampi.Citation1 These findings suggest that the Aβ-abundant brain homogenate of Alzheimer disease patients may possess the ability to induce or supplement the overproduction of Aβ, possibly leading to the onset of Alzheimer disease.
The pathological feature associated with Parkinson disease is the formation of Lewy bodies in cell bodies and neuronal processes in the brain.Citation2 The main component of these protein aggregates is α-synuclein (reviewed in ref. Citation2). Autopsies of Parkinson disease patients revealed that Lewy bodies had formed on healthy embryonic neurons that had been grafted onto the brain tissue of the patients several years before (prior to said examination).Citation3–Citation5 It may thus be proposed that α-synuclein transmission is possible from diseased to healthy neurons, suggesting that Parkinson disease may be transmissible from a Parkinson disease patient to a healthy individual. These findings imply that Alzheimer and Parkinson diseases may be transmissible through tissue transplants and the use of contaminated surgical tools.Citation6
Prion disorders, also termed Transmissible Spongiform Encephalopathies (TSEs), are fatal neurodegenerative diseases that affect the central nervous system (CNS) of multiple animal species. In lieu of the social, economic and political ramifications of such infections, as well as the possible intra- and interspecies transmissibility of such disorders, various routes of experimental transmission have been investigated including intracerebral, intraperitoneal, intraventricular, intraocular, intraspinal and subcutaneous injections (reviewed in ref. Citation7–Citation9). However, such routes of transmission are not representative of the “natural” mechanism as the majority of prion disorders are contracted through ingestion of infectious prion (PrPSc) containing material. Thus, the peroral and perenteral prion transmission is of greatest consequence with respect to TSE disease establishment. Moreover, the presence of PrPSc in the buccal cavity of scrapie-infected sheepCitation10 (reviewed in ref. Citation11) and the possible horizontal transfer as a result hereof, as may be similarly proposed for animals suffering from other TSEs, may further contribute to the oral transmissibility of TSEs.
A number of model systems have been employed to study TSE transmissibility. Owing to ethical constraints, TSE transmissibility to humans via the oral route may not be directly investigated and as a result hereof, alternative model systems are needed. These may include the use of transgenic mice, cell lines which are permissive to infectionCitation12 and experimental animals such as sheep, calves, goats, minks, ferrets and non-human primates (reviewed in ref. Citation9).
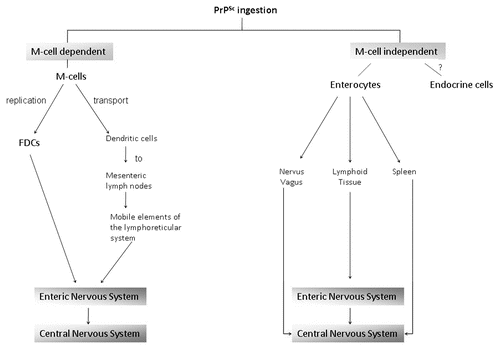
Intestinal entry of PrPSc has been proposed to occur via two pathways, the membranous (M) cell-dependent and M cell-independent pathways ().Citation13,Citation14 The former involves endocytic M (microfold)-cells, which cover the intestinal lymphoid follicles (Peyer's patches)Citation14 and may take up prions and thereby facilitate the translocation of these proteins across the intestinal epithelium into the lymphoid tissues (reviewed in ref. Citation9) as has been demonstrated in a cellular model.Citation13 Following such uptake by the M cells, the prions may subsequently pass to the dendritic cells and follicular dendritic cells (FDCs) (), which allow for prion transport to the mesenteric lymph nodes and replication, respectively.Citation15 The prion proteins may subsequently gain access to the enteric nervous system (ENS) and ultimately the central nervous system (CNS).Citation15
However, prion intestinal translocation has been observed in the absence of M cells and has been demonstrated to be as a result of the action of polar, 37 kDa/67 kDa LRP/LR (non-integrin laminin receptor; reviewed in ref. Citation16–Citation18) expressing enterocytes. Enterocytes are the major cell population of the intestinal epithelium and due to their ability to endocytose pathogens, nutrients and macromolecules,Citation19 it has been proposed that these cells may represent a major entry site for alimentary prions ().
Since enterocyte prion uptake has been demonstrated to be dependent on the presence of LRP/LR on the apical brush border of the cells,Citation14,Citation20 the interaction between varying prion protein strains and the receptorCitation21–Citation23 may be employed as a model system to study possible oral transmissibility of prion disorders across species as well as the intestinal pathophysiology of alimentary prion infections.Citation24 Moreover, the blockage of such interactions through the use of anti-LRP/LR specific antibodies has been reported to reduce PrPSc endocytosisCitation19 and thus these antibodies may serve as potential therapeutics to prevent infectious prion internalization and thereby prevent prion infections. It must be emphasized that the adhesion of prion proteins to cells is not solely dependent on the LRP/LR-PrPSc interactions;Citation24 however, this interaction is of importance with regards to internalization and subsequent pathogenesis.
We applied the aforementioned cell model to study the possible oral transmission of PrPBSE, PrPCWD and ovine PrPSc to cervids, cattle, swine and humans.Citation24 The direct transmission of the aforementioned animal prion disorders to humans as a result of dietary exposure and the possible establishment of zoonotic diseases is of great public concern. It must however be emphasized that the study investigated the co-localization of LRP/LR and various prion strains and not the actual internalization process.
PrPBSE was shown to co-localize with LRP/LR on human enterocytesCitation24, thereby suggesting that PrPBSE is transmissible to humans via the oral route which is widely accepted as the manner by which variant CJD originated. This suspicion was previously investigated using a macaque model, which was successfully perorally infected by BSE-contaminated material and subsequently lead to the development of a prion disorder that resembles vCJD.Citation25 These results, due to the evolutionary relatedness between macaques and humans, allowed researchers to confirm the oral transmissibility of PrPBSE to humans. PrPBSE may also potentially lead to prion disorder establishment in swine,Citation24 livestock of great economic and social importance.
The prion disorder affecting elk, mule deer and white-tailed deer is termed CWD. Cases of the disease are most prevalent in the US but are also evident in Canada and South Korea.Citation26,Citation27 As the infectious prion isoform is reported to be present in the bloodCitation28 and skeletal muscle,Citation29 hunting, consumption of wild venison and contact with other animal products derived from CWD-infected elk and deer may thereby pose a public health risk. Our studies demonstrate that PrPCWD co-localizes with LRP/LR on human enterocytesCitation24 thereby suggesting a possible oral transmissibilty of this TSE to humans. This is, however, inconsistent with results obtained during intra-cerebral inoculation of the brains and spinal cords of transgenic mice overexpressing the human cellular prion protein (PrPc),Citation26,Citation27 which is essential for TSE disease establishment and progression. Further, discrepancies have also been reported with respect to non-human primates, as squirrel monkeys have been successfully intracerebrally inoculated with mule-deer prion homogenates,Citation30 while cynolmolgus macaques were resistant to infection.Citation31 CWD has been transmitted to ferrets, minks and goatsCitation32 and as these animals may serve as domestic animals or livestock, secondary transmission from such animals to humans, through direct contact or ingestion of infected material, may be an additional risk factor that merits further scientific investigation.
Ovine PrPSc co-localization with LRP/LR on human and bovine enterocytes may be indicative of the infectious agents' ability to effect cross-species infections. The oral transmissibility of Scrapie has been confirmed in hamsters fed with sheep-scrapie-infected material.Citation33
The discrepancies with regards to the transmissibility of certain infectious prion proteins when assessed by different model systems may be due to the experimental transmission route employed. Oral exposure often results in significantly prolonged incubation times when compared to intracerebral inoculation techniques and thus failure of transgenic mice and normal experimental animals to develop disease phenotypes after being fed TSE-contaminated material may not necessarily indicate that the infection process failed.Citation14 Apart from the route of infection, numerous other factors may influence transmission between species, including dose, PrP polymorphisms and genetic factors, the prion strain employed as well as the efficacy of prion transport to the CNS.Citation34 The degree of homology between the PrPc protein in the animals serving as the infectious prion source and recipient has also been described as a feature limiting cross-species transmission.Citation34 The negative results, as referred to above, obtained upon prion-protein inoculation of animal models may have resulted due to the slow rate at which the infectious prion induces conformational conversion of the endogenous PrPc in the animal cells and this in turn results in low levels of infectious prion replication and symptom development.Citation27
Furthermore, even in the event that certain prion disorders are not directly transmissible to humans, most are transmissible to at least a single species of domestic animal or livestock. The infectious agents properties may be altered in the secondary host such that it becomes transmissible to humans (reviewed in ref. Citation35). Thus, interspecies transmission between animals may indirectly influence human health.
It is noteworthy to add that although the oral route of PrPSc transmission may result in prolonged incubation times, it may broaden the range of susceptible hosts. A common constituent of food is ferritin, a protein that is resistant to digestive enzyme hydrolysis and, due to its homology across species, it may serve as co-transporter of PrPSc and facilitate enterocyte internalization of the infectious prion.Citation36 It may thus be proposed that prion internalization may occur via a ferritin-PrPSc complex even in the absence of co-localization between the infectious agent and LRP/LR such that many more cross-species infections (provided that the other infection factors are favorable) may be probable.Citation37 In addition, digestive enzymes in the gastrointestinal tract facilitate PrPSc binding to the intestinal epithelium and subsequent intestinal uptakeCitation36 and thus depending on the individuals' digestive processes, the susceptibility to infection and the rate of disease development may vary accordingly. As a result hereof, though laboratory experiments in cell-culture and animal models may render a particular prion disorder non-infectious to humans, this may not be true for all individuals.
In lieu of the above statements, with particular reference to inconsistencies in reported results and the multiple factors influencing oral transmissibility of TSEs, further transmission studies are required to evaluate the zoonotic threat which CWD, BSE and Scrapie may pose through ingestion.
Abbreviations
| PrPc | = | cellular prion protein |
| PrPSc | = | scrapie prion protein |
| TSE | = | transmissible spongiform encephalopathy |
| LRP/LR | = | non-integrin laminin receptor |
| M-cells | = | microfold cells |
| FDCs | = | follicular dendritic cells |
| CJD | = | Creutzfeldt-Jakob disease |
| BSE | = | Bovine Spongiform Encephalopathy |
| CWD | = | Chronic Wasting disease |
| ENS | = | enteric nervous system |
| CNS | = | central nervous system |
| Aβ | = | amyloid-β |
Figures and Tables
Figure 1 Proposed routes of gastrointestinal entry of ingested infectious prions (PrPSc) as well as possible pathways of amplification and transport to the central nervous system.
Acknowledgements
We thank the National Research Foundation (NRF), Republic of South Africa for financial support.
References
- Meyer-Luehmann M, Coomaraswamy J, Bolmont T, Kaeser S, Schaefer C, Kilger E, et al. Exogenous induction of cerebral beta-amyloidogenesis is governed by agent and host. Science 2006; 313:1781 - 1784
- Olanow CW, Tatton WG. Etiology and pathogenesis of Parkinson's disease. Annu Rev Neurosci 1999; 22:123 - 144
- Kordower JH, Brundin P. Lewy body pathology in long-term fetal nigral transplants: is Parkinson's disease transmitted from one neural system to another?. Neuropsychopharmaco 2009; 34:254
- Kordower JH, Chu Y, Hauser RA, Freeman TB, Olanow CW. Lewy body-like pathology in long-term embryonic nigral transplants in Parkinson's disease. Nat Med 2008; 14:504 - 506
- Kordower JH, Chu Y, Hauser RA, Olanow CW, Freeman TB. Transplanted dopaminergic neurons develop PD pathologic changes: a second case report. Mov Disord 2008; 23:2303 - 2306
- Brundin P, Melki R, Kopito R. Prion-like transmission of protein aggregates in neurodegenerative diseases. Nat Rev Mol Cell Biol 2010; 11:301 - 307
- Kimberlin RH, Cole S, Walker CA. Pathogenesis of scrapie is faster when infection is intraspinal instead of intracerebral. Microb Pathog 1987; 2:405 - 415
- Kimberlin RH, Walker CA. Pathogenesis of scrapie (strain 263K) in hamsters infected intracerebrally, intraperitoneally or intraocularly. J Gen Virol 1986; 67:255 - 263
- Weissmann C, Enari M, Klohn PC, Rossi D, Flechsig E. Transmission of prions. J Infect Dis 2002; 186:157 - 165
- Maddison BC, Rees HC, Baker CA, Taema M, Bellworthy SJ, Thorne L, et al. Prions are secreted into the oral cavity in sheep with preclinical scrapie. J Infect Dis 2010; 201:1672 - 1676
- Da Costa Dias B, Weiss SF. A kiss of a prion: new implications for oral transmissibility. J Infect Dis 2010; 201:1615 - 1616
- Neale MH, Mountjoy SJ, Edwards JC, Vilette D, Laude H, Windl O, et al. Infection of cell lines with experimental and natural ovine scrapie agents. J Virol 2010; 84:2444 - 2452
- Ghosh S. Mechanism of intestinal entry of infectious prion protein in the pathogenesis of variant Creutzfeldt-Jakob disease. Adv Drug Deliv Rev 2004; 56:915 - 920
- Shmakov AN, Ghosh S. Prion proteins and the gut: une liaison dangereuse?. Gut 2001; 48:443 - 447
- Davies GA, Bryant AR, Reynolds JD, Jirik FR, Sharkey KA. Prion diseases and the gastrointestinal tract. Can J Gastroenterol 2006; 20:18 - 24
- Mbazima V, Da Costa Dias B, Omar A, Jovanovic K, Weiss SFT. Interaction between PrPc and other ligands with the 37 kDa/67 kDa laminin receptor. Front Biosci 2010; 15:1150 - 1163
- Vana K, Zuber C, Pflanz H, Kolodziejczak D, Zemora G, Bergmann AK, et al. LRP/LR as an alternative promising target in therapy of prion diseases, Alzheimer's disease and cancer. Infect Disord Drug Targets 2009; 9:69 - 80
- Omar A, Jovanovic K, Da Costa Dias B, Gonsalves D, Moodley K, Caveney R, et al. Patented biological approaches for the therapeutic modulation of the 37 kDa/67 kDa laminin receptor. Expert Opin Ther Pat 2011; 21:35 - 53; http://dx.doi.org/10.1517/13543776.2011.539203
- Morel E, Andrieu T, Casagrande F, Gauczynski S, Weiss S, Grassi J, et al. Bovine prion is endocytosed by human enterocytes via the 37 kDa/67 kDa laminin receptor. Am J Pathol 2005; 167:1033 - 1042
- Shmakov AN, Bode J, Kilshaw PJ, Ghosh S. Diverse patterns of expression of the 67 kD laminin receptor in human small intestinal mucosa: potential binding sites for prion proteins?. J Pathol 2000; 191:318 - 322
- Rieger R, Edenhofer F, Lasmezas CI, Weiss S. The human 37 kDa laminin receptor precursor interacts with the prion protein in eukaryotic cells. Nat Med 1997; 3:1383 - 1388
- Gauczynski S, Nikles D, El-Gogo S, Papy-Garcia D, Rey C, Alban S, et al. The 37 kDa/67 kDa laminin receptor acts as a receptor for infectious prions and is inhibited by polysulfated glycanes. J Infect Dis 2006; 194:702 - 709
- Gauczynski S, Peyrin JM, Haik S, Leucht C, Hundt C, Rieger R, et al. The 37 kDa/67 kDa laminin receptor acts as the cell-surface receptor for the cellular prion protein. Embo J 2001; 20:5863 - 5875
- Kolodziejczak D, Da Costa Dias B, Zuber C, Jovanovic K, Omar A, Beck J, et al. Prion interaction with the 37 kDa/67 kDa laminin receptor on enterocytes as a cellular model for intestinal uptake of prions. J Mol Biol 2010; 402:293 - 300
- Lasmezas CI, Deslys JP, Demaimay R, Adjou KT, Lamoury F, Dormont D, et al. BSE transmission to macaques. Nature 1996; 381:743 - 744
- Sandberg MK, Al-Doujaily H, Sigurdson CJ, Glatzel M, O'Malley C, Powell C, et al. Chronic wasting disease prions are not transmissible to transgenic mice overexpressing human prion protein. J Gen Virol 2010; 91:2651 - 2657
- Tamguney G, Giles K, Bouzamondo-Bernstein E, Bosque PJ, Miller MW, Safar J, et al. Transmission of elk and deer prions to transgenic mice. J Virol 2006; 80:9104 - 9114
- Mathiason CK, Powers JG, Dahmes SJ, Osborn DA, Miller KV, Warren RJ, et al. Infectious prions in the saliva and blood of deer with chronic wasting disease. Science 2006; 314:133 - 136
- Angers RC, Browning SR, Seward TS, Sigurdson CJ, Miller MW, Hoover EA, et al. Prions in skeletal muscles of deer with chronic wasting disease. Science 2006; 311:1117
- Marsh RF, Kincaid AE, Bessen RA, Bartz JC. Interspecies transmission of chronic wasting disease prions to squirrel monkeys (Saimiri sciureus). J Virol 2005; 79:13794 - 13796
- Race B, Meade-White K, Race R, Chesebro B. Prion infectivity in fat of deer with chronic wasting disease. J Virol 2009; 83:9608 - 9610
- Bartz JC, Marsh RF, McKenzie DI, Aiken JM. The host range of chronic wasting disease is altered on passage in ferrets. Virology 1998; 251:297 - 301
- McBride PA, Beekes M. Pathological PrP is abundant in sympathetic and sensory ganglia of hamsters fed with scrapie. Neurosci Lett 1999; 265:135 - 138
- Raymond GJ, Bossers A, Raymond LD, O'Rourke KI, McHolland LE, Bryant PK 3rd, et al. Evidence of a molecular barrier limiting susceptibility of humans, cattle and sheep to chronic wasting disease. EMBO J 2000; 19:4425 - 4430
- Belay ED, Maddox RA, Williams ES, Miller MW, Gambetti P, Schonberger LB. Chronic wasting disease and potential transmission to humans. Emerg Infect Dis 2004; 10:977 - 984
- Mishra RS, Basu S, Gu Y, Luo X, Zou WQ, Mishra R, et al. Protease-resistant human prion protein and ferritin are cotransported across Caco-2 epithelial cells: implications for species barrier in prion uptake from the intestine. J Neurosci 2004; 24:11280 - 11290
- Sunkesula SR, Luo X, Das D, Singh A, Singh N. Iron content of ferritin modulates its uptake by intestinal epithelium: implications for co-transport of prions. Mol Brain 2010; 3:14