Abstract
The relationship between Alzheimer disease (AD) and prion-related encephalopathies (TSE) has been proposed by different points of view. Recently, the scientific attention has been attracted by the results proposing the possibility that PrPc, the protein whose pathologic form is responsible of TSE, can mediated the toxic effect of β amyloid (Aβ) oligomers. The oligomers are considered the culprit of the neurodegenerative process associated to AD, although the pathogenic mechanism activated by these small aggregates remain to be elucidated. In the initial study based on the binding screening PrPc was identified as ligand /receptor of Aβ oligomers, while long term potentiation (LTP) analysis in vitro and behavioural studies in vivo, demonstrated that the absence of PrPc abolished the damage induced by Aβ oligomers. The high affinity binding Aβ oligomers-PrPc has been confirmed, whereas a functional role of this association has been excluded by three different studies. We approached this issue by the direct application of Aβ oligomers in the brain followed by the behavioural examination of memory deficits. Our data using PrP knock-out mice suggest that Aβ 1-42 oligomers are responsible for cognitive impairment in AD but PrPc is not required for their effect. Similarly, in two other studies the LTP alterations induced by Aβ 1-42 oligomers was not influenced by the absence of PrP. Possible explanations of these contradictory results are discussed.
AD is the main cause of dementia in elderly, with prevalence that doubles every five years of age from the 0.5% at 65 years old. AD is characterized by the cerebral accumulation of Aβ in extracellular plaques and the formation of intracellular neurofibrillary tangles constituted by hyperphosphorylated tau protein; the two pathological features are sequentially associated.Citation1 TSE are rare transmissible neurodegenerative disorders with heterogeneous phenotypes,Citation2 the genetic cases are associated with mutations of prion protein genes.Citation3 The conformational change from the cellular (PrPc) to the pathological (PrPSc) form of prion protein characterized the TSE and PrPSc is considered the infectious agent that propagates by autocatalytic mechanism in the host.Citation4 The presence of PrP mutations favors the shift from PrPc to PrPSc. Both AD and TSE belong to the group of neurodegener-ative disorders associated to protein misfolding. A common molecular mechanism has been proposed to explain the pathogenesis of these disorders characterized by the extracellular accumulation of misfolded proteins. Despite the enormous differences in terms of incidence (ten case AD/103/year, one TSE/106/year, 10,000-fold less) genetic and neuropathological studies have proposed possible associations in specific populations or in single subjects between AD and TSE. Several evidences indicate that polymorphism at residue 129, a key element in TSE, might also influence the susceptibility to AD.Citation5–Citation7 More recently, a direct interaction between Aβ and prion protein has been proposed by several points of view. The studies by Hooper's group reported that PrPc can influence the metabolism of amyloid precursor protein (APP) and the production of Aβ, (reviewed in ref. Citation8). A series of proteolytic passages mediated the formation of Aβ from APP, the amyloidogenic pathway is initiated by the cleavage by β secretase (BACE) producing soluble βAPP and the C-terminal C99 membrane bound fragment, whereas the following cleavage mediated by γ secretase complex forms Aβ and the amyloid intracellular domain (AICD). Alternatively, the non-amyloidogenic pathway α secretase cleaves APP at in the middle of Aβ sequence forming soluble αAPP and C-terminal fragment. Multiple peptidases, including neprysil, endothelium-converting enzyme and insulin degrading enzyme are involved in Aβ degradation. The accumulation of Aβ is consequence of an imbalance between the formation and the degradation of the peptide. Apparently PrPc can influence this mechanism through a reduction of β secretase cleavage. The initial results were produced in a human neuroblastoma cell line where the overexpression of PrPc was associated with a reduction of Aβ production, whereas when PrPc was silenced by siRNA the Aβ levels increased. An increase of Aβ was also observed in PrP knockout mice due to a reduction in APP cleavage by BACE. Further investigations by the same authors indicate that PrPc can interact directly with BACE when both are localized in lipid raft, where β secretase cleavage preferentially occurs and glycosaminoglycans played an essential role in mediating this interaction. A regulation of PrP expression by AICD fragment is suggested by the data of Vincent et al.Citation9 where the traslocation of this fragment acting as transcription factor interacting with the promoter of PrP. Based on this evidence, Kellett and HooperCitation8 proposed a potential feedback loop between AICD and PrPc. The production of AICD and Aβ is inhibited by PrPc by the modulation of β secretase, in turn an excess of AICD activated the PrPc synthesis. Thus, the physiological production of Aβ appears controlled by PrPc. This mechanism is interesting but its effective role in vivo is controversial. Recently Calella et al.Citation10 showed that double APP/PS1 transgenic mice Aβ and APP levels were not altered by the different expression of PrPc. However, in this case the presence of PS1 mutation might alter the metabolic pathway associated to Aβ production and the PrPc is no more effective.
A crosstalk between misfolded proteins was proposed by Morales et al.Citation11 In this study it has been shown that the inoculation of experimental scrapie in transgenic mice overexpressing human APP (Tg2576) showed an acceleration of onset of clinical symptoms and a shortening of survival time, modest but significant, compared to wild-type littermate. However, neither the histopathological profile of TSE lesions nor the strain feature of the inoculum (RML) was found altered by the passage in Tg 2576 mice. On the other hand, an increase of brain inflammation and Aβ deposition was observed in Tg2576 animals inoculated with experimental scrapie. Furthermore, the determination of PrPSc at the same post inoculation days in transgenic or wild-type mice showed a clear increase accordingly with the anticipation of symptoms. Based also on the cross seeding demonstrated in vitro using synthetic Aβ and recombinant PrP, the authors explained the synergistic effect in the two diseases as consequence of a direct interaction between the two proteins favoring the protein misfolding. Heterogeneous seeding between different amyloidogenic proteins has been shown by in vitro and in vivo findings;Citation12,Citation13 this can occur also in the human diseases under appropriate conditions.
The more attractive finding on the interaction between Aβ and PrPc came from the evidence shown by Lauren et al.Citation14 indicating PrPc as a, or “the,” receptor capable to mediate the neurotoxic effect of Aβ oligomers. Oligomers, soluble small aggregates of Aβ, are considered responsible for synaptic and cognitive dysfunction as well as neurodegenerative effect in AD. When the neurotoxicity of Aβ peptides was initially described, it was strictly associated to their fibrillogenic capacity;Citation15,Citation16 however, we found that the toxicity of Aβ peptide could be independent of its aggregation state.Citation17 Successively, the studies of Walsh and Selkoe demonstrated that protofibrils and oligomers rather than fibers can be responsible of neurodegeneration in AD.Citation18 In agreement with this concept is the strong correlation between the synaptic loss with cortical levels of soluble Aβ species rather than with plaques distribution in AD patients.Citation18–Citation20 In vitro and in vivo studies have now indicated that soluble Aβ oligomers impair synaptic plasticity, inhibiting hippocampal LTP, the electrophysiological correlate of synaptic plasticity.Citation21–Citation23 Memory impairment and LTP inhibition have also been detected in AD mouse models before plaque deposition in the brain parenchyma.Citation24–Citation26 The mechanism through which Aβ oligomers act remains uncertain, but interactions have been reported with several receptors such as nicotinic, insulinic and glutamatergic receptors, leading to detrimental effects on synaptic plasticity and spine formation.Citation27–Citation29 Thus, using an oligomeric preparation according to Klein's group (ADDL),Citation30,Citation31 Lauren et al.Citation14 identified the PrPc as Aβ oligomers ligand based on the screening of cDNA expression library of more than 200,000 proteins. The authors further confirmed these results by the evaluation of the affinity of PrPc for Aβ oligomers in various conditions. The functional consequence of this association was investigated in vitro by the electrophysological determination LTP suppression induced by Aβ oligomers, in PrP knockout cells, this inhibition was completely abolished. While the binding of Aβ oligomers to PrPc is a result foreseen because PrPc has hydrophobic domains that can attract the oligomers, specific interaction with functional consequence was not obvious. Using surface plasmon resonance (SPR) technique and in vivo experimental model we investigated the ability of PrPc to bind Aβ oligomers and its involvement in their action.Citation32 Several types of Aβ aggregates isolated from biological sources have been used in rats to test the involvement of Aβ oligomers in memory impairment.Citation33 In our study we determined which Aβ assemblies are responsible for memory deficit in C57BL/6 receiving into lateral ventricle infusions of initial state, oligomers or fibrils of synthetic Aβ1–42 and assessed their cognitive performance in the novel object recognition task.Citation34,Citation35 We demonstrated that synthetic Aβ oligomers can induce an immediate memory impairment in mice; the effect was detectable at a nanomolar concentration of Aβ oligomers (10–50 nM). In the same conditions, freshly solubilized Aβ (manly monomeric form) or fibrils were not active. In addition, a 10-day later retest without further oligomer injection found that the memory performance was fully restored showing that the deficit was transient. When we distinguished the effects of Aβ oligomers on memory encoding/consolidation or retrieval we found that they inhibited the encoding/consolidation of information, without affecting its retrieval if properly stored. In fact, the i.c.v. injection of Aβ oligomers before acquisition of the information during the familiarization phase prevented the information being either encoded or consolidated. In contrast, when the Aβ oligomers were applied only 24 h after the information had been processed, no deficit was detected, suggesting that Aβ oligomers do not abolish the retrieval of stabilized information, but do prevent its encoding or consolidation. Memory processing requires NMDA receptor activation and intracellular signaling leading to AMPA receptor trafficking, synthesis of new proteins and formation of dendritic spines.Citation36–Citation38 In another study using APP23 transgenic mice we found direct evidence that electrophysiological function mediated by NMDA receptors was altered before the formation of Aβ plaques.Citation39 Thus, our findings indicate the interaction with NMDA receptors as responsible of the detrimental activity of Aβ oligomers.
When Aβ oligomers were injected i.c.v. in PrP knockout mice, we found that the effect of Aβ oligomers on memory was preserved and comparable to that of wild-type mice. We also tested the involvement of PrPC in mediating Aβ oligomer toxicity in vitro, by investigating their effect on survival of primary hippocampal neurons from wild type or Prnp0/0 cells. Aβ oligomers were toxic to both Prnp+/+ and Prnp0/0 hippocampal cells, consistent with the conclusion that their adverse effects are independent of PrPC. In the past we already published results comparing PrP 106–126 neurotoxicity with the toxic effect of Aβ 25–35 in Prnp0/0 hippocampal cells, the effect of PrP 106–126 disappeared while the toxicity of Aβ fragment was similar to that found in wild-type neurons.Citation40 On the other hand, surface plasmon resonance (SPR) confirmed a high-affinity interaction between PrPc and Aβ oligomers. PrPc from mouse brain homogenates was captured on the sensor surface of SPR chips by either 3F4 or 94B4, two anti-PrPc antibodies. The captured protein was actually PrPc since no signal was detected when flowing brain homogenate coming from Prnp0/0 mice. PrPc captured by both 94B4 and 3F4 maintains its ability to bind 6D11, an anti-PrP antibody directed against the epitope 93–109, i.e., the region suggested to be involved in the interaction with Aβ oligomers. In these conditions Aβ oligomers bound PrPc specifically, while Aβ initial state and fibrils did not. These data were confirmed by Chen et al.Citation41 who specifically analyzed the binding of Aβ oligomers to PrPc using both SPR and side-directed spin labeling. These authors found that in addition to the previously postulated 95–110 region, the segment 23–27 of PrPc is critically important for effective binding to Aβ oligomers. The affinity of synthetic Aβ for recombinant PrP was confirmed by Calella et al.Citation10 using less sophisticated technique. The functional role of Aβ oligomers/PrPc interaction was further investigated by Strittmatter's group crossing PrP0/0 mice with APP/PS1 transgenic mice.Citation42 The memory deficit exhibited by APP/PS1 aged mice was completely rescued when the animals were grown on PrP0/0 background. The APP/PS1/PrP0/0 mice not only performed better cognitively but also had a significant reduction of mortality compared to APP/PS1/PrP+/+. According to these results the absence of PrPc was sufficient to avoid the cognitive deficits, presumably due to Aβ oligomers, and to improve the general conditions of an AD animal model. In a collaborative study, the same group recently reported that a treatment with an anti-prion antibody for two weeks can also complete nullified the cognitive impairment observed in the APP/PS1 transgenic mice.Citation43 However two other papers did not confirm the influence of PrPc on Aβ oligomers activity. Calella et al.Citation10 showed that either the ablation or the overexpression of PrPc had no effect on the impairment of hippocampal LTP in double APP/PS1 transgenic mice. The suppression of LTP found in APP/PS1 mice was not altered when PrP was overexpressed or nullified, importantly these results were obtained with a particular attention to minimize the influence of the genetic background of the mice used. Finally, Kessels et al.Citation44 using two different methods, showed that Aβ1–42 oligomers mediated LTP suppression was independent from the presence of PrPc. Organotypic hippocampal slice neurons infected with a virus that increased the production and secretion of Aβ showed a depression of synaptic activity in both wild-type or PrP0/0 slices. Similar effect in both conditions was found when a reduction of neuronal spines was induced by the APP overexpression or direct application of Aβ1–42 oligomers in organotypic hippocampal slices. The authors replicated the LTP inhibition induced by Aβ1–42 oligomers but this effect in their hands was not influenced by the absence of PrPc.
Taken together these results are in agreement on two relevant points, first, the capacity of oligomers deriving from synthetic Aβ1–42 peptide, not only bio-derived preparation as suggested by others,Citation23 to induce an impairment of synaptic plasticity and the memory deficits both in vitro and in vivo. Second point, the Aβ1–42 oligomers do exhibit a high affinity for PrPc, as originally shown by Lauren et al.Citation14 and the 95–110 domain is involved in this binding capacity. The discrepancies emerge when we consider functional aspects consequent to the physical interaction between Aβ1–42 oligomers and PrPC, relevant and consistent for one group, poorly or inexistent for the others. Differences in methodological approaches could explain these conflicting results although the consistency of the other two aspects such as Aβ1–42 oligomer detrimental action and their binding to PrPC, would argue against this possible explanation. As suggested by Lauren et al.Citation45 in their reply, the different electrophysiological results in vitro, could be for instance consequence of the different Aβ1–42 oligomers concentration used. To rule out this possibility, in our experimental conditions we tested either our original preparation of Aβ1–42 oligomers, obtained with an incubation of Aβ1–42 peptide overnight at 4°C and the Aβ1–42 oligomers preparation obtained following the procedure described by Lauren et al.Citation14 Accordingly to the atomic force microscopy (AFM) analysis the peptide solution at 22°C included not only oligomers but also few other species, fibers and protofibers, while at 4°C only spherical oligomers were detectable. However, the two preparations exerted similar effects either in the SPR binding studies or in the behavioral tests, thus suggesting that in our hands oligomer concentration and the presence of other chemicophysical species did not influence the biological effect. We must, however, take into account that if small changes in the methodological conditions can nullified the role by PrPc as mediator of the Aβ1–42 oligomer detrimental action on synaptic function, the relevance of the evidence becomes negligible. On the other hand, however the results obtained in APP/PS1/PrP0/0 mice, where the absence of PrPc can completely rescue the behavioral deficits, are in favor of a central role of PrPc in Aβ1–42 oligomers toxicity. As mentioned above the PrP0/0 background was shown to affect also the survival of APP/PS1 transgenic mice indicating a favorable influence apparently behind the simple interaction with Aβ1–42 oligomers activity. Furthermore, as raised by Peretti,Citation46 both APP and PS1 transgene in the mice used by Strittmatter's group are under the control of prion promoter. This make the Aβ production and the presence of PrPc strictly associated over the physiological conditions. Several aspects thus remain to be clarify before to drive any firm conclusion. The interaction between Aβ1–42 oligomers and PrPC has been tested in a single experimental model, and the positive effects of PrPC ablation in transgenic mice were observable only at twelve months of age when amyloid plaques were already detectable. For this reason, the use of different strains is essential to confirm the relevance of PrPc in vivo. For instance, in APP23 transgenic mice we have shown that memory deficits occurred before the deposition of Aβ in plaques.Citation39 A possible explanation of these results is that soluble oligomers inducing memory deficits are formed and they actively affect the cognitive behavior in an early pathological phase that precedes the plaques formation. An association between the appearance of oligomers and memory deficits was found by Lesne et al.Citation29 while profibrils formation and behavioral deficits before plaque formation have been shown in transgenic mice combined Arctic and Swedish mutation.Citation47 If this interpretation is correct, it is assumable that the ablation of PrPC and, consequently its binding to Aβ oligomers, would abolish the cognitive decline also in these conditions. The appropriate controls are also necessary for the treatment with anti-PrP antibody in APP/PS1 mice, such as the treatment with an antibody that recognizes a domain of PrP not involved in the binding with Aβ1–42 oligomers.
However the main problem raised by the functional role of PrPc as mediator of oligomer toxic effects is the unconvincing concept that PrPc is responsible for more that 50% of Aβ1–42 oligomers binding to neuronal membrane. A recent study has shown that initial exposure of Aβ1–42 oligomers, within one hour, affect the membrane mobility of metabotropic glutamate receptors (mGluR5). The Aβ1–42 oligomers induced a clustering of mGluR5 in hippocampal cells that elevate the intracellular calcium, potentially responsible of synaptic deterioration.Citation48 Similar effect was obtained with artificial crosslinking of mGluR5 that increased the intracellular calcium and promote synaptotoxicity. According to this study the interaction of Aβ1–42 oligomers with mGluR5 is specific, and the application of an antibody against the extracellular domain of these receptors reduced the binding of oligomers of about 40%. Interestingly, the reduction of Aβ1–42 oligomer binding after incubation with the anti-prion antibody (6D11) was also confirmed in this study, but when the antibodies anti-prion and anti-mGluR5 were applied together no increase of the effect with respect to the single antibody was observed. Even with the addition of a third antibody directed against NR1, NMDA receptor subunit, that also reduced the binding of Aβ1–42 oligomers by itself, but the total effect did not go over the 30% of binding reduction. Although the incubation time of this experiment was very brief (15 min), no further reduction of the binding was found. The proximity of the different antigens with the Aβ1–42 oligomer binding site might explain the result.
On the other hand, the hippocampal cells cultured from mGluR50/0 mice showed a reduction of the Aβ1–42 oligomers binding sites in the neuronal spines of about 80%, in contrast with the findings of Lauren et al.Citation14, which showed in PrP0/0 a 60% reduction of the oligomers binding sites. Several other proteins have been proposed as receptors for Aβ1–42 oligomer, such as the voltage-gated calcium channel,Citation49 the angiotensin II receptorCitation50 or the nicotinic receptors.Citation51,Citation52 Furthermore, a new concept of a multi-component assembly required for stabilizing the toxic accumulation of Aβ oligomers at the synaptic membrane involving NMDA receptors has been recently proposed.Citation53 Taken together, these results indicate that the quantification of Aβ1–42 oligomer binding sites is approximate and the relevance of one or the other potential receptors is strongly influenced by the research conditions used.
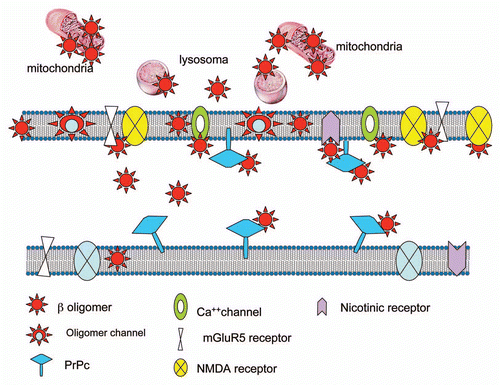
Furthermore, robust evidence indicates that the interaction between oligomers and cellular membrane can occur without a specific mediation of any proteic entity.Citation54–Citation56 The Aβ1–42 oligomers are inserted in the membrane with change in the membrane fluidity and alteration of citoarchitecture or even internalized within the cells. Several findings have demonstrated that Aβ oligomers are directly incorporated into neuronal membranes and form calcium-permeable ion channels (amyloid channels).Citation57,Citation58 This concept, originally proposed by Arispe et al.Citation59, is based on the measurement of ionic current through artificial bilayer membrane exposed to Aβ1–42 peptide. Similar results were obtained also with neurotoxic prion peptides 106–126 and 82–146.Citation60,Citation61 In a recent paper a structural model of oligomers constituted by tetrameric and hexameric β-sheet subunits has been proposed on the basis of chemicophysical and biochemical evidence.Citation62 Along this line a potential therapeutic approach has been proposed based on the possibility to block with small molecules the amyloid channel and consequently to antagonize Aβ oligomers neurotoxicity.Citation58 Using an original cellular model, other authors have recently shown that the disruption of lipid rafts affect the toxicity of Aβ oligomers.Citation63 Using proteomic and immunocytochemistry approaches we found that Aβ toxic species crossed the plasma membrane and accumulated in cells through binding to a variety of internal proteins.Citation64 In neuroblastoma cell line, Aβ oligomers interact with a large number of proteins located on the cytoskeleton and in the endoplasmatic reticulum. On the other hand, an accumulation of Aβ oligomers was found in the mitochondrial compartment of APP transgenic mice supporting the growing evidence of a major role of mitochondria in AD pathogenesisCitation65 and underlining the capacity of Aβ oligomers to activate an intraneuronal neurotoxic pathway. In conclusion, the molecular mechanisms responsible for Aβ oligomers synaptotoxicity is extremely complex and the interaction with the neuronal membrane can occur at various levels with multiple processes (). In specific conditions it is possible that PrPc might exert a relevant role, probably as consequence of a sequestration of Aβ oligomers rather than a functional activity directly associated to the protein. Taken together, all this evidence makes us conclude that we are still very far away from proving the absolute relevance of this phenomenon in AD pathogenesis.
Abbreviations
| AD | = | Alzheimer disease |
| ADDLs | = | amyloid β-derived diffusible ligand |
| Aβ | = | β amyloid |
| AFM | = | atomic force microscopy |
| AICD | = | amyloid intracellular domain |
| APP | = | amyloid precursor protein |
| BACE | = | β secretase |
| LTP | = | long term potentation |
| mGLuR5 | = | metabotropic glutamate receptors |
| PrPc | = | cellular prion protein |
| PrPSc | = | pathological form of prion protein |
| RML | = | Rocky Mountain Laboratory strain |
| SPR | = | surface plasmon resonance |
| TSE | = | transmissible spongiform encephalopathies |
Figures and Tables
Figure 1 Multiple interactions of Aβ oligomers with different cellular structures.
References
- Querfurth HW, LaFerla FM. Alzheimer's disease. N Engl J Med 2010; 362:329 - 344
- Collins SJ, Lawson VA, Masters CL. Transmissible spongiform encephalopathies. Lancet 2004; 363:51 - 61
- Mastrianni JA, Brown K. The prion diseases. J Geriatr Psychiatry Neurol 2010; 23:277 - 298
- DeArmond SJ, Prusiner SB. Perspectives on prion biology, prion disease pathogenesis and pharmacologic approaches to treatment. Clin Lab Med 2003; 23:1 - 41
- Del Bo R, Scarlato M, Ghezzi S, Martinelli-Boneschi F, Fenoglio C, Galimberti G, et al. Is M129V of PRNP gene associated with Alzheimer's disease? A case-control study and a meta-analysis. Neurobiol Aging 2006; 27:770
- Gacia M, Safranow K, Styczynska M, Jakubowska K, Peplonska B, Chodakowska-Zebrowska M, et al. Prion protein gene M129 allele is a risk factor for Alzheimer's disease. J Neural Transm 2006; 113:1747 - 1751
- Giannattasio C, Poleggi A, Puopolo M, Pocchiari M, Antuono P, Dal Forno G, et al. Survival in Alzheimer's disease is shorter in women carrying heterozygosity at codon 129 of the PRNP gene and no APOE epsilon 4 allele. Dement Geriatr Cogn Disord 2008; 25:354 - 358
- Kellett KA, Hooper NM. Prion protein and Alzheimer disease. Prion 2009; 3:190 - 194
- Vincent B, Sunyach C, Orzechowski HD, St. George-Hyslop P, Checler F. p53-dependent transcriptional control of cellular prion by presenilins. J Neurosci 2009; 29:6752 - 6760
- Calella AM, Farinelli M, Nuvolone M, Mirante O, Moos R, Falsig J, et al. Prion protein and Abeta-related synaptic toxicity impairment. EMBO Mol Med 2010; 2:306 - 314
- Morales R, Estrada LD, Diaz-Espinoza R, Morales-Scheihing D, Jara MC, Castilla J, et al. Molecular cross talk between misfolded proteins in animal models of Alzheimer's and prion diseases. J Neurosci 2010; 30:4528 - 4535
- Jean L, Thomas B, Tahiri-Alaoui A, Shaw M, Vaux DJ. Heterologous amyloid seeding: revisiting the role of acetylcholinesterase in Alzheimer's disease. PLoS ONE 2007; 2:652
- Vishveshwara N, Liebman SW. Heterologous cross-seeding mimics cross-species prion conversion in a yeast model. BMC Biol 2009; 7:26
- Laurén J, Gimbel DA, Nygaard HB, Gilbert JW, Strittmatter SM. Cellular prion protein mediates impairment of synaptic plasticity by amyloid-beta oligomers. Nature 2009; 457:1128 - 1132
- Lorenzo A, Yankner BA. Beta-amyloid neurotoxicity requires fibril formation and is inhibited by congo red. Proc Natl Acad Sci USA 1994; 91:12243 - 12247
- Pike CJ, Walencewicz-Wasserman AJ, Kosmoski J, Cribbs DH, Glabe CG, Cotman CW. Structure-activity analyses of beta-amyloid peptides: contributions of the beta 25–35 region to aggregation and neurotoxicity. J Neurochem 1995; 64:253 - 265
- Forloni G, Lucca E, Angeretti N, Della Torre P, Salmona M. Amidation of beta-amyloid peptide strongly reduced the amyloidogenic activity without alteration of the neurotoxicity. J Neurochem 1997; 69:2048 - 2054
- Walsh DM, Selkoe DJ. A beta oligomers—a decade of discovery. J Neurochem 2007; 101:1172 - 1184
- Lue LF, Kuo YM, Roher AE, Brachova L, Shen Y, Sue L, et al. Soluble amyloid beta peptide concentration as a predictor of synaptic change in Alzheimer's disease. Am J Pathol 1999; 155:853 - 886
- McLean CA, Cherny RA, Fraser FW, Fuller SJ, Smith MJ, Beyreuther K, et al. Soluble pool of Abeta amyloid as a determinant of severity of neurodegeneration in Alzheimer's disease. Ann Neurol 1999; 46:860 - 886
- Näslund J, Haroutunian V, Mohs R, Davis KL, Davies P, Greengard P, Buxbaum JD. Correlation between elevated levels of amyloid beta-peptide in the brain and cognitive decline. JAMA 2000; 283:1571 - 1578
- Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M, et al. Diffusible, nonfibrillar ligands derived from Abeta1–42 are potent central nervous system neurotoxins. Proc Natl Acad Sci USA 1998; 95:6448 - 6453
- Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, et al. Amyloid-beta protein dimers isolated directly from Alzheimer's brains impair synaptic plasticity and memory. Nat Med 2008; 14:837 - 842
- Townsend M, Shankar GM, Mehta T, Walsh DM, Selkoe DJ. Effects of secreted oligomers of amyloid beta-protein on hippocampal synaptic plasticity: a potent role for trimers. J Physiol 2006; 572:477 - 492
- Wang HW, Pasternak JF, Kuo H, Ristic H, Lambert MP, Chromy B, et al. Soluble oligomers of beta amyloid (1–42) inhibit long-term potentiation but not long-term depression in rat dentate gyrus. Brain Res 2002; 924:133 - 140
- Hsia AY, Masliah E, McConlogue L, Yu GQ, Tatsuno G, Hu K, et al. Plaque-independent disruption of neural circuits in Alzheimer's disease mouse models. Proc Natl Acad Sci USA 1999; 96:3228 - 3233
- Mucke L, Masliah E, Yu GQ, Mallory M, Rockenstein EM, Tatsuno G, et al. High-level neuronal expression of abeta 1–42 in wild-type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. J Neurosci 2000; 20:4050 - 4058
- Cleary JP, Walsh DM, Hofmeister JJ, Shankar GM, Kuskowski MA, Selkoe DJ, et al. Natural oligomers of the amyloid-beta protein specifically disrupt cognitive function. Nat Neurosci 2005; 8:79 - 84
- Lesne S, Koh MT, Kotilinek L, Kayed R, Glabe CG, Yang A, et al. A specific amyloid-beta protein assembly in the brain impairs memory. Nature 2006; 440:352 - 357
- Chromy BA, Nowak RJ, Lambert MP, Viola KL, Chang L, Velasco P, et al. Self-assembly of Abeta(1–42) into globular neurotoxins. Biochemistry 2003; 42:12749 - 12760
- Klein WL. Abeta toxicity in Alzheimer's disease: globular oligomers (ADDLs) as new vaccine and drug targets. Neurochem Int 2002; 41:345 - 352
- Balducci C, Beeg M, Stravalaci M, Bastone A, Sclip A, Biasini E, et al. Synthetic amyloid-beta oligomers impair long-term memory independently of cellular prion protein. Proc Natl Acad Sci USA 2010; 107:2295 - 2300
- Ashe KH, Zahs KR. Probing the biology of Alzheimer's disease in mice. Neuron 2010; 66:631 - 645
- Huang SM, Mouri A, Kokubo H, Nakajima R, Suemoto T, Higuchi M, et al. N Neprilysin-sensitive synapse-associated amyloid-beta peptide oligomers impair neuronal plasticity and cognitive function. J Biol Chem 2006; 281:17941 - 17945
- Shankar GM, Bloodgood BL, Townsend M, Walsh DM, Selkoe DJ, Sabatini BL. Natural oligomers of the Alzheimer amyloid-beta protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J Neurosci 2007; 27:2866 - 2875
- Collingridge GL, Isaac JT, Wang YT. Receptor trafficking and synaptic plasticity. Nat Rev Neurosci 2004; 5:952 - 962
- Luscher C, Nicoll RA, Malenka RC, Muller D. Synaptic plasticity and dynamic modulation of the postsynaptic membrane. Nat Neurosci 2000; 3:545 - 550
- Hsieh H, Boehm J, Sato C, Iwatsubo T, Tomita T, Sisodia S, et al. AMPAR removal underlies Abeta-induced synaptic depression and dendritic spine loss. Neuron 2006; 52:831 - 843
- Balducci C, Tonini R, Zianni E, Nazzaro C, Fiordaliso F, Salio M, et al. Cognitive deficits associated with alteration of synaptic metaplasticity precede plaque deposition in APP23 transgenic mice. J Alzheimers Dis 2010; 21:1367 - 1381; http://dx.doi.org/10.3233/JAD-2010-100675
- Fioriti L, Quaglio E, Massignan T, Colombo L, Stewart RS, Salmona M, et al. The neurotoxicity of prion protein (PrP)peptide 106–126 is independent of the expression level of PrP and is not mediated by abnormal PrP species. Mol Cell Neurosci 2005; 28:165 - 176
- Chen S, Yadav SP, Surewicz WK. Interaction between human prion protein and amyloid-beta (Abeta) oligomers: role of N-terminal residues. J Biol Chem 2010; 285:26377 - 26383
- Gimbel DA, Nygaard HB, Coffey EE, Gunther EC, Laurén J, Gimbel ZA, et al. Memory impairment in transgenic Alzheimer mice requires cellular prion protein. J Neurosci 2010; 30:6367 - 6374
- Chung E, Ji Y, Sun Y, Kascsak RJ, Kascsak RB, Mehta PD, et al. Anti-PrPC monoclonal antibody infusion as a novel treatment for cognitive deficits in an alzheimer's disease model mouse. BMC Neurosci 2010; 11:130
- Kessels HW, Nguyen LN, Nabavi S, Malinow R. The prion protein as a receptor for amyloid-beta. Nature 2010; 466:3 - 4
- Laurén J, Gimbel DA, Nygaard HB, Gilbert JW, Strittmatter SM. Reply. Nature 2010; 466:4 - 5
- Peretti D. Is PrPC a mediator of Aâ toxicity in Alzheimer's disease?. J Neurosci 2010; 30:11883 - 11884
- Lord A, Englund H, Söderberg L, Tucker S, Clausen F, Hillered L, et al. Amyloid-beta protofibril levels correlate with spatial learning in Arctic Alzheimer's disease transgenic mice. FEBS J 2009; 276:995 - 1006
- Renner M, Lacor PN, Velasco PT, Xu J, Contractor A, Klein WL, et al. Deleterious effects of amyloid beta oligomers acting as an extracellular scaffold for mGluR5. Neuron 2010; 66:739 - 754
- Nimmrich V, Grimm C, Draguhn A, Barghorn S, Lehmann A, Schoemaker H, et al. Amyloid beta oligomers (Abeta(1–42) globulomer) suppress spontaneous synaptic activity by inhibition of P/Q-type calcium currents. J Neurosci 2008; 28:788 - 797
- Abdalla S, Lother H, el Missiry A, Langer A, Sergeev P, el Faramawy Y, et al. Angiotensin II AT2 receptor oligomers mediate G-protein dysfunction in an animal model of Alzheimer disease. J Biol Chem 2009; 284:6554 - 6565
- Dougherty JJ, Wu J, Nichols RA. Beta-amyloid regulation of presynaptic nicotinic receptors in rat hippocampus and neocortex. J Neurosci 2003; 23:6740 - 6747
- Liu Q, Huang Y, Xue F, Simard A, DeChon J, Li G, et al. A novel nicotinic acetylcholine receptor subtype in basal forebrain cholinergic neurons with high sensitivity to amyloid peptides. J Neurosci 2009; 29:918 - 929
- Decker H, Jürgensen S, Adrover MF, Brito-Moreira J, Bomfim TR, Klein WL, et al. N-methyl-D-aspartate receptors are required for synaptic targeting of Alzheimer's toxic Aβ oligomers. J Neurochem 2010; 115:1520 - 1529
- Deshpande A, Mina E, Glabe C, Busciglio J. Different conformations of amyloid beta induce neurotoxicity by distinct mechanisms in human cortical neurons. J Neurosci 2006; 26:6011 - 6018
- Stefani M. Biochemical and biophysical features of both oligomer/fibril and cell membrane in amyloid cytotoxicity. FEBS J 2010; 277:4602 - 4613
- Sepulveda FJ, Parodi J, Peoples RW, Opazo C, Aguayo LG. Synaptotoxicity of Alzheimer beta amyloid can be explained by its membrane perforating property. PLoS One 2010; 5:11820
- Kawahara M. Neurotoxicity of amyloid protein: oligomerization, channel formation and calcium dyshomeostasis. Curr Pharm Des 2010; 16:2779 - 2789
- Diaz JC, Simakova O, Jacobson KA, Arispe N, Pollard HB. Small molecule blockers of the Alzheimer Abeta calcium channel potently protect neurons from A beta cytotoxicity. Proc Natl Acad Sci USA 2009; 106:3348 - 3353
- Arispe N, Rojas E, Pollard HB. Alzheimer disease amyloid β-protein forms calcium channels in bilayer membranes: Blockade by tromethamine and aluminum. Proc Natl Acad Sci USA 1993; 90:567 - 571
- Kourie JI, Kenna BL, Tew D, Jobling MF, Curtain CC, Masters CL, et al. Copper modulation of ion channels of PrP[106–126] mutant prion peptide fragments. J Membr Biol 2003; 193:35 - 45
- Bahadi R, Farrelly PV, Kenna BL, Kourie JI, Tagliavini F, Forloni G, et al. Channels formed with a mutant prion protein PrP(82–146) homologous to a 7 kDa fragment in diseased brain of GSS patients. Am J Physiol Cell Physiol 2003; 285:862 - 872
- Strodel B, Lee JW, Whittleston CS, Wales DJ. Transmembrane structures for Alzheimer's Aβ(1–42) oligomers. Am Chem Soc 2010; 132:13300 - 13312
- Malchiodi-Albedi F, Contrusciere V, Raggi C, Fecchi K, Rainaldi G, Paradisi S, et al. Lipid raft disruption protects mature neurons against amyloid oligomer toxicity. Biochim Biophys Acta 2010; 1802:406 - 415
- Manzoni C, Colombo L, Messa M, Cagnotto A, Balducci C, Forloni G, et al. Membrane proteins and Aβ peptide toxicity. Alzheimers Dement 2009; 5:490 - 491
- Galindo MF, Ikuta I, Zhu X, Casadesus G, Jordán J. Mitochondrial biology in Alzheimer's disease pathogenesis. J Neurochem 2010; 114:933 - 945