Abstract
Misfolded proteins are at the core of many neurodegenerative diseases, nearly all of them associated with cognitive impairment. For example Creutzfeldt-Jacob disease is associated with aggregation of prion protein,1,2 Lewy body dementia and Parkinson disease with alpha-synuclein3,4
Currently, the diagnosis of AD is based on exclusion of other forms of impairment with definitive diagnosis requiring autopsy confirmation.Citation30 Thus, there is a strong need to find easily measurable in vivo AD biomarkers that could facilitate early and accurate diagnosisCitation31 as well as prognostic data to assist in monitoring therapeutic efficacy.Citation32 Although biological markers such as MRI, PET scans and CSF increase the diagnostic likelihood that AD is present,Citation9,Citation18–Citation20,Citation33,Citation34 biomarkers are invasive, uncomfortable, expensive and may not be readily available to rural areas, underserved communities, underinsured individuals or developing countries, making them impractical for broad use. However, the lessons learned from biomarkers can be applied to increase the likelihood that clinicians will be able to detect disease at earlier stages in the form of dementia screening.
Public health may be best defined as the organized efforts of society to improve health, often framed in terms of primary, secondary and tertiary prevention. Prevention encompasses an understanding of causation, alteration of natural history of disease and understanding of pathophysiological mechanisms.Citation35 The clearest application of this from a public health perspective is in the setting of secondary prevention (i.e., screening)—early detection as a core element, coupled with treatments or preventative actions to reduce the burden of disease.Citation35 In this instance we seek to identify individuals in whom a disease has already begun and who may be experiencing very mild clinical symptoms but have not yet sought out medical care. The objective of effective screening is to detect the disease earlier than it would have been detected with usual care. Recent healthcare reform (Accountable Care Act)Citation36 proposes a Personalized Prevention Plan including screening for cognitive disorders, reimbursable through Medicare. Thus tying knowledge about dementia screening with underlying biology of protein misfolding associated with neurodegenerative disease can have enormous implications.
A review of the natural history of dementia illustrates this point (). The timeline of disease from presumptive start to the patient demise is plotted. Stage I marks the biologic onset of disease; however this point often cannot be identified and may begin years to decades before any evidence is apparent (represented by dashed lines). As this stage is subclinical, it is difficult to study in humans but lends itself nicely to animal models. At some point in the progression of the biology, stage II begins heralding the first pathologic evidence of disease could be obtained—in the case of AD this could include CSF measurements of amyloid and tauCitation22,Citation26,Citation27 or PET imaging with amyloid ligands.Citation18,Citation37 Subsequently, the first signs and symptoms of disease develop (stage III). Till this point, the disease process has been entirely presymptomatic. Beginning with the onset of symptoms, the patient may seek medical care (stage IV) and eventually be diagnosed (stage V). From stage III onwards, the patient enters the symptomatic phase of disease. From this point, the patient is typically treated with various pharmacologic and nonpharmacologic approaches towards some outcome. Another way to envision the disease spectrum is from the biological onset to the seeking of medical attention as the preclinical phase of disease with the clinical phase beginning with the initial clinical investigations into the cause of the patients' symptoms.
What is the value of thinking about disease in this fashion? Such models allow researchers and clinicians to model the approach to finding and applying new diagnostics and offering new interventions. From stage I to stage III, the patient is the presymptomatic, preclinical phase of disease. The only means of detection would be with a biological marker that reflected protein misfolding or some proxy marker of these events. Although longitudinal evidence of cognitive change exist from 1–3 years before clinical diagnosis, raw scores on neuropsychological testing during this time remains in the normal range.Citation38 After stage IV, the patient is in the symptomatic, clinical phase of disease. Testing here is centered on confirming the suspected diagnosis, correctly staging the disease and initiating the appropriate therapies. Basic scientific approaches focusing on the presymptomatic, preclinical phase and clinical care approaches focusing on the symptomatic, clinical phase are well established and will continue to benefit from additional research.
However, if we focus only on these two phases, an opportunity will be missed to make a decidedly important impact in the patient's well-being. From stage III to stage IV, the patient enters symptomatic, preclinical phase of disease; symptomatic because the patient or family is beginning to detect some aspect of change, but preclinical because these signs and symptoms have not yet been brought to medical attention. In the case of AD (and the other forms of dementia) this period may go for an extended length of time as patients, families and clinicians dismiss early cognitive symptoms as part of the normal aging process. Thus, the rationale for screening is that if we can identify disease earlier in its natural history than would ordinarily occur, intervention measures (those currently available and those that are being developed) would be more effective. Dementia screening therefore would be best suited to detect cognitive impairment at the beginning of disease signs (stage III), particularly if these screening measures reflect what is known about the symptomatic, clinical phase of disease and correlate with the pathologic changes occurring in the brain during the pre-symptomatic, preclinical phase of disease.
In a recent paper, we evaluated the relationship between several dementia screening tests and biomarkers of AD.Citation40 We tested whether a reliable and validated informant-based dementia screening test (the AD8)Citation41,Citation42 correlates with changes in AD biomarkers and, if positive, screening with the AD8 clinically supports an AD clinical phenotype, superior to a commonly used performance-based screening tests including the Mini Mental State Exam (MMSE)Citation43 and the Short Blessed Test (SBT).Citation44 A total of 257 participants were evaluated, administered a comprehensive clinical and cognitive evaluation with the Clinical Dementia Rating scale (CDR)Citation45 used as the gold standard. Participants consented to and completed a variety of biomarker studies including MRI, amyloid imaging using the Pittsburgh Compound B (PiB)Citation37,Citation46 and CSF studies of Aβ42, tau and phosphorylated tau at Serine 181 (p-tau181).Citation23,Citation24 The sample had a mean age of 75.4 ± 7.3 years with 15.1 ± 3.2 years of education. The sample was 88.7% Caucasian and 45.5% male with a mean MMSE score of 27.2 ± 3.6. The formal diagnoses of the sample was 156 CDR 0 cognitively normal, 23 CDR 0.5 MCI, 53 CDR 0.5 very mild AD and 25 CDR 1 mild AD. Participants with positive AD8 scores (graded as a score of 2 or greater) exhibited the typical AD fluid biomarker phenotype characterized by significantly lower mean levels of CSF Aβ42, greater CSF tau, p-tau181 and the tau(s)/Aβ42 ratios.Citation26,Citation27 They also exhibited smaller temporal lobe volumes and increased mean cortical binding potential (MCBP) for PiB imaging similar to studies of individuals with AD.Citation18,Citation19 These findings support that informant-based assessments may be superior to performance-based screening measures such as the MMSE or SBT in corresponding to underlying AD pathology, particularly at the earliest stages of decline. The use of a brief test such as the AD8 may improve strategies for detecting dementia in community settings where biomarkers may not be readily available and also may enrich clinical trial recruitment by increasing the likelihood that participants have underlying biomarker abnormalities.Citation40
To gain a better understanding of changes in biomarkers in the symptomatic, preclinical phase, a post hoc evaluation of the 156 individuals who were rated as CDR 0 no dementia at the time of their Gold Standard assessment was completed. Some of these nondemented individuals have abnormal AD biomarkers, but in the absence of performing lumbar punctures or PET scans, is it possible to detect evidence of change? AD8 scores for 132 individuals were less than 2; thus their screening test suggests no impairment (mean AD8 score = 0.30 ± 0.46). However 25 of these individuals had AD8 scores (≥2) suggesting impairment (mean AD8 score = 2.4 ± 0.91). Applying the model described in , some of these individuals are hypothesized to be in the symptomatic, preclinical phase of disease. No difference in age, education, gender or brief performance tests (MMSE or SBT) were detected between groups (); however, the CDR sum of boxesCitation45 is increased in the individuals with higher AD8 scores supporting that informants were noticing and reporting changes in the participants cognitive function. A review of the individual AD8 questions that were first reported to change suggest that informants endorsement of subtle changes in memory (repeats questions, forgets appointments) and executive ability (trouble with judgment, appliances, finances) are valuable early signs. This is consistent with previous reports that changes in memory and judgment/problem solving CDR boxscores in nondemented individuals correlate with findings of AD pathology at autopsy.Citation17 Although biomarkers do not reach significance in this small sample, the direction of change in favor of “Alzheimerization” of this group suggests that some of these individuals may be in the symptomatic, preclinical phase of disease. More research with larger sample sizes and longitudinal follow-up is needed to confirm this hypothesis. It should be also noted that not all individuals with an AD8 score of 2 or greater have AD. The AD8 was designed to detect cognitive impairment from all causes, and as such, these mildly affected individuals may have other causes for their cognitive change such as depression, Lewy body dementia or vascular cognitive impairment.Citation41,Citation42
To explore this further, changes in AD biomarkers (CSF Aβ42, Tau and PiB-PET) were plotted against the age of the participant (). Previous research suggest that biomarker changes are more commonly seen in older populationsCitation47 and increasing age is the greatest risk factor for developing AD.Citation7 AD8 scores of 0 or 1 (no impairment) are depicted as filled circles while AD8 scores of 2 or greater (impairment) are depicted as open squares. Regression lines are plotted for the entire cohort (dashed black line) and for each subset (black for AD8 no impairment; gray for AD8 Impairment). The top row (Parts A–C) represents biomarker profiles for the entire sample of 257 individuals divided by their AD8 scores. With age, there are changes in biomarkers with decreasing CSF Aβ42 (A), increasing CSF Tau (B) and increased PiB-PET binding potential (C). The effect of age on CSF biomarkers is most marked in the AD8 No Impairment group (black line) while changes in PiB binding is seen only in the AD8 Impaired group (gray line). The second row in (Parts D–F) represents biomarker profiles for the 156 individuals who were rated as CDR 0 no dementia at the time of their Gold Standard, 25 of whom had AD8 scores in the impaired range. Some of these individuals are hypothesized to be in the symptomatic, preclinical phase of AD. Similar age-related changes in CSF Aβ42 and PiB binding are seen with CSF Aβ42 having the greatest rate of decline in the AD8 no impairment group and PiB binding having the greatest rate of change in the AD8 impairment group. Increases in CSF Tau are seen as a function of age regardless of group.
While a number of interpretations are possible from this type of data, if one considers the model of disease in it appears that CSF changes in Aβ42 and Tau precede PiB binding changes in the presymptomatic, preclinical phase of disease consistent with previous attempts at modeling AD.Citation25 Even with sensitive measurements, this phase is unlikely to be detected without some biological evaluation. At the start of the symptomatic, preclinical phase of AD, PiB binding increases and this may be detected by careful evaluation of the patient and a knowledgeable informant with a validated dementia screening instrument such as the AD8. As patients move into the symptomatic, clinical phase of disease, biomarkers are markedly abnormal as is most cognitive testing permitting careful staging and prognostication.
AD and related disorders will become a public health crisis and a severe burden on Medicare in the next two decades unless actions are taken to (1) develop disease modifying medications,Citation48 (2) provide clinicians with valid and reliable measures to detect disease at the earliest possible stage and (3) reimburse clinicians for their time to do so. While this perspective does not address development of new therapeutics, it should be clear that regardless of what healthcare reform in the US eventually looks like,Citation1 dementia screening is a viable means to detect early disease as it enters its symptomatic phase. Dementia screening with the AD8 offers the additional benefit of corresponding highly with underlying disease biology of AD that includes alteration of protein conformation, protein misfolding and eventual aggregation of these misfolded proteins as plaques and tangles.
Financial Disclosure
Drs. James E. Galvin, Catherine M. Roe and John C. Morris hold the copyright for the AD8.
Financial Support
J.E.G.'s time was supported by P30 AG008051 from the National Institute on Aging, National Institutes of Health.
Figures and Tables
Figure 1 Model of the natural history of AD. Timeline from presumptive start of AD through patient diagnosis is plotted. The initiation of biological changes (stage I) marks the onset of disease and begins years to decades before any evidence is apparent (represented by dashed lines). At some point the first pathologic evidence of disease (stage II) begins and in theory can be detected with biomarkers such as CSF measurements of amyloid and tau or PET imaging with amyloid ligands. Subsequently, the first signs and symptoms of disease develop (stage III) followed by the patient seeking medical attention (stage IV) and finally a diagnosis is established (stage V). This timeline can be clustered into a presymptomatic phase (stages I–III) and a symptomatic phase (stages III–V). An alternative way to envision the disease spectrum is from the biological onset to the seeking of medical attention (stages I–IV) as the preclinical phase of disease with the clinical phase beginning with the initial clinical investigations into the cause of the patients' symptoms (stages IV and V). Stage III is the ideal time for dementia screening.
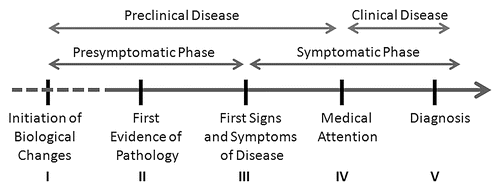
Figure 2 Changes in AD biomarkers by age and AD8 scores. AD biomarkers are plots as a function of age (x-axis) and AD8 scores. AD8 scores of 0 or 1 (no impairment) are depicted as filled circles while AD8 scores of 2 or greater (impairment) are depicted as open squares. Regression lines are plotted for the entire cohort (dashed black line) and for each subset (black for AD8 no impairment; gray for AD8 impairment). The top row (A–C) represents biomarker profiles for the entire cohort (n = 257) divided by their AD8 scores. With age, there are changes in biomarkers with decreasing CSF Aβ42 (A), increasing CSF Tau (B) and increased PiB-PET binding potential (C). The effect of age on CSF biomarkers is most marked in the AD8 no impairment group (black line) while changes in PiB binding is seen only in the AD8 impaired group (gray line). The bottom row (D–F) represents biomarker profiles for the individuals rated CDR 0 no dementia (n = 156), 25 of whom had AD8 scores in the impaired range. Similar age-related changes in CSF Aβ42 and PiB binding are seen with CSF Aβ42 having the greatest rate of decline in the AD8 no impairment group and PiB binding having the greatest rate of change in the AD8 impairment group. Increases in CSF Tau are seen as a function of age regardless of group.
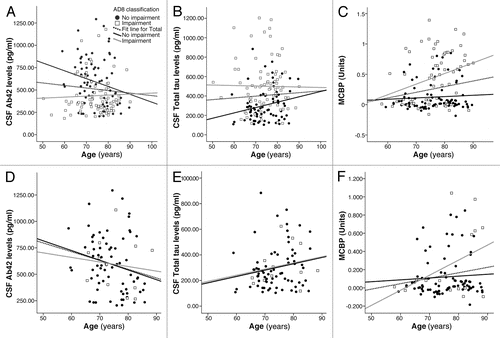
Table 1 Characteristics of nondemented CDR 0 individuals stratified by AD8 scores
Acknowledgements
The author would like to thank the Clinical, Biomarker, Neuroimaging and Genetics Cores of the Washington University Alzheimer Disease Research Center (P50 AG05681, J.C. Morris, Principle Investigator) for the clinical, cognitive, ApoE genotyping and biomarker assessments. The development and validation of the AD8 was supported by grants P01 AG03991, P01 AG026276 and P50 AG05681 from the National Institute on Aging, National Institutes of Health (J.C. Morris, PI) and the Longer Life Foundation. A copy of the AD8 and scoring instructions may be viewed at alzheimer.wustl.edu/About_Us/PDFs/AD8form2005.pdf.
References
- Venneti S. Prion diseases. Clin Lab Med 2010; 30:293 - 309
- Sharma S, Mukherjee M, Kedage V, Muttigi MS, Rao A, Rao S. Sporadic Creutzfeldt-Jakob disease—a review. Int J Neurosci 2009; 119:1981 - 1994
- Tarawneh R, Galvin JE. Distinguishing Lewy body dementias from Alzheimer's disease. Expert Rev Neurother 2007; 7:1499 - 1516
- Tan JM, Wong ES, Lim KL. Protein misfolding and aggregation in Parkinson's disease. Antioxid Redox Signal 2009; 11:2119 - 2134
- Bugiani O. The many ways to frontotemporal degeneration and beyond. Neurol Sci 2007; 28:241 - 244
- Cairns NJ, Ghoshal N. FUS: A new actor on the frontotemporal lobar degeneration stage. Neurology 2010; 74:354 - 356
- Alzheimer Association. Facts and Figures 2010; 11/15/10 http://www.alz.org
- Petersen RC, Roberts RO, Knopman DS, Boeve BF, Geda YE, Ivnik RJ, et al. Mild cognitive impairment: ten years later. Arch Neurol 2009; 66:1447 - 1455
- Vemuri P, Wiste HJ, Weigand SD, Knopman DS, Trojanowski JQ, Shaw LM, et al. Alzheimer's Disease Neuroimaging Initiative: Serial MRI and CSF biomarkers in normal aging, MCI and AD. Neurology 2010; 75:143 - 151
- Funke SA, Birkmann E, Willbold D. Detection of Amyloid-beta aggregates in body fluids: a suitable method for early diagnosis of Alzheimer's disease?. Curr Alzheimer Res 2009; 6:285 - 289
- Galvin JE. Alzheimer disease: Understanding the challenges, improving the outcome. App Neurol 2007; 3 - 13
- Galvin JE. Alzheimer's disease: Diagnosis and treatment across the spectrum. App Neurol 2005; 1:1 - 16
- Perrin RJ, Fagan AM, Holtzman DM. Multimodal techniques for diagnosis and prognosis of Alzheimer's disease. Nature 2009; 461:916 - 922
- Hampel H, Shen Y, Walsh DM, Aisen P, Shaw LM, Zetterberg H, et al. Biological markers of amyloid beta-related mechanisms in Alzheimer's disease. Exp Neurol 2010; 223:334 - 346
- Hampel H, Bürger K, Teipel SJ, Bokde AL, Zetterberg H, Blennow K. Core candidate neurochemical and imaging biomarkers of Alzheimer's disease. Alzheimers Dement 2008; 4:38 - 48
- Price JL, McKeel DW Jr, Buckles VD, Roe CM, Xiong C, Grundman M, et al. Neuropathology of nondemented aging: presumptive evidence for preclinical Alzheimer disease. Neurobiol Aging 2009; 30:1026 - 1036
- Galvin JE, Powlishta KK, Wilkins K, McKeel DW Jr, Storandt M, Grant E, et al. Predictors of preclinical Alzheimer disease and dementia: a clinicopathologic study. Arch Neurol 2005; 62:758 - 765
- Mintun MA, LaRossa GN, Sheline YI, Dence CS, Lee SY, Mach RH, et al. [11C] PIB in a nondemented population: potential antecedent marker of Alzheimer disease. Neurology 2006; 67:446 - 452
- Morris JC, Roe CM, Grant EA, Head D, Storandt M, Goate AM, et al. Pittsburgh compound B imaging and prediction of progression from cognitive normality to symptomatic Alzheimer disease. Arch Neurol 2009; 66:1469 - 1475
- De Meyer G, Shapiro F, Vanderstichele H, Vanmechelen E, Engelborghs S, De Deyn PP, et al. Alzheimer's Disease Neuroimaging Initiative. Diagnosis-independent Alzheimer disease biomarker signature in cognitively normal elderly people. Arch Neurol 2010; 67:949 - 956
- Blennow K, Hampel H, Weiner M, Zetterberg H. Cerebrospinal fluid and plasma biomarkers in Alzheimer disease. Nat Rev Neurol 2010; 6:131 - 144
- Mattsson N, Zetterberg H, Hansson O, Andreasen N, Parnetti L, Jonsson M, et al. CSF biomarkers and incipient Alzheimer disease in patients with mild cognitive impairment. JAMA 2009; 302:385 - 393
- Fagan AM, Mintun MA, Shah AR, Aldea P, Roe CM, Mach RH, et al. Cerebrospinal fluid tau and ptau(181) increase with cortical amyloid deposition in cognitively normal individuals: implications for future clinical trials of Alzheimer's disease. EMBO Mol Med 2009; 1:371 - 380
- Fagan A, Mintun M, Mach R, Lee SY, Dence CS, Shah AR, et al. Inverse relation between in vivo amyloid imaging load and CSF Abeta42 in humans. Ann Neurol 2006; 59:512 - 519
- Jack CR Jr, Knopman DS, Jagust WJ, Shaw LM, Aisen PS, Weiner MW, et al. Hypothetical model of dynamic biomarkers of the Alzheimer's pathological cascade. Lancet Neurol 2010; 9:119 - 128
- Shaw LM, Vanderstichele H, Knapik-Czajka M, Clark CM, Aisen PS, Petersen RC, et al. Alzheimer's Disease Neuroimaging Initiative: Cerebrospinal fluid biomarker signature in Alzheimer's disease neuroimaging initiative subjects. Ann Neurol 2009; 65:403 - 413
- Fagan AM, Roe CM, Xiong C, Mintun MA, Morris JC, Holtzman DM. Cerebrospinal fluid tau/β-amyloid42 ratio as a prediction of cognitive decline in nondemented older adults. Arch Neurol 2007; 64:343 - 349
- Hampel H, Broich K. Enrichment of MCI and early Alzheimer's disease treatment trials using neurochemical and imaging candidate biomarkers. J Nutr Health Aging 2009; 13:373 - 375
- Mattsson N, Zetterberg H, Blennow K. Lessons from multicenter studies on CSF biomarkers for Alzheimer's disease. Int J Alzheimers Dis 2010; 610 - 613
- McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer's disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer's Disease. Neurology 1984; 34:939 - 944
- Dubois B, Feldman HH, Jacova C, Dekosky ST, Barberger-Gateau P, Cummings J, et al. Research criteria for the diagnosis of Alzheimer's disease: revising the NINCDS-ADRDA criteria. Lancet Neurol 2007; 6:734 - 746
- Urbanelli L, Magini A, Ciccarone V, Trivelli F, Polidoro M, Tancini B, et al. New perspectives for the diagnosis of Alzheimer's disease. Recent Pat CNS Drug Discov 2009; 4:160 - 181
- Berti V, Osorio RS, Mosconi L, Li Y, De Santi S, de Leon MJ. Early detection of Alzheimer's disease with PET imaging. Neurodegener Dis 2010; 7:131 - 135
- Brys M, Glodzik-Sobanska L, Switalski R, Rich K, Pirraglia E, Mosconi L, et al. MRI Improves CSF biomarkers in the early detection of Alzheimer's disease. J Alz Dis 2008; 16:351 - 362
- Wright CF, Hall A, Matthews FE, Brayne C. Biomarkers, dementia and public health. Ann NY Acad Sci 2009; 1180:11 - 19
- HR2959: Accountable Care Promotion Act of 2009 11/15/10 www.govtrack.us/congress/bill.xpd?bill=h111-2959
- Klunk WE, Engler H, Nordberg A, Wang Y, Blomqvist G, Holt DP, et al. Imaging brain amyloid in Alzheimer's disease with Pittsburgh Compound-B. Ann Neurol 2004; 55:306 - 319
- Haldenwanger A, Eling P, Kastrup A, Hildebrandt H. Correlation between cognitive impairment and CSF biomarkers in amnesic MCI, non-amnesic MCI and Alzheimer's disease. J Alzheimers Dis 2010; 22:971 - 980
- Johnson DK, Storandt M, Morris JC, Galvin JE. Longitudinal study of the transition from healthy aging to Alzheimer disease. Arch Neurol 2009; 66:1254 - 1259
- Galvin JE, Fagan AM, Holtzman DM, Mintun MA, Morris JC. Relationship of dementia screening tests with biomarkers of Alzheimer's Disease. Brain 2010; 133:3290 - 3300
- Galvin JE, Roe C, Coats M, Powlishta KK, Muich SJ, Grant E, et al. The AD8: A brief informant interview to detect dementia. Neurology 2005; 65:559 - 564
- Galvin JE, Roe CM, Xiong C, Morris JC. The validity and reliability of the AD8 informant interview for dementia. Neurology 2006; 67:1942 - 1948
- Folstein MF, Folstein SE, McHugh PR. Mini-mental State: A practical method for grading the cognitive state of patients for the clinicians. J Psychiatr Res 1975; 12:189 - 198
- Katzman R, Brown T, Fuld P, Peck A, Schechter R, Schimmel H. Validation of a short orientation-memory-concentration test of cognitive impairment. Am J Psychiatry 1983; 140:734 - 739
- Morris JC. The clinical dementia rating (CDR): current version and scoring rules. Neurology 1993; 43:2412 - 2414
- Mathis CA, Wang Y, Holt DP, Huang GF, Debnath ML, Klunk WE. Synthesis and evaluation of 11C-labeled 6-substituted 2-arylbenzothiazoles as amyloid imaging agents. J Med Chem 2003; 46:2740 - 2754
- Glodzik-Sobanska L, Pirraglia E, Brys M, De Santi S, Mosconi L, Rich KE, et al. The effects of normal aging and ApoE genotype on the levels of CSF biomarkers for Alzheimer's disease. Neurobiol Aging 2009; 30:672 - 681
- Tarawneh R, Galvin JE. Potential future neuroprotective therapies for neurodegenerative disorders and stroke. Clinic Geriatr Med 2010; 26:125 - 147