Abstract
The biochemical essence of prion replication is the molecular multiplication of the disease-associated misfolded isoform of prion protein (PrP), termed PrPSc, in a nucleic acid-free manner. PrPSc is generated by the protein misfolding process facilitated by conformational conversion of the host-encoded cellular PrP to PrPSc. Evidence suggests that an auxiliary factor may play a role in PrPSc propagation. We and others previously discovered that plasminogen interacts with PrP, while its functional role for PrPSc propagation remained undetermined. In our recent in vitro PrP conversion study, we showed that plasminogen substantially stimulates PrPSc propagation in a concentration-dependent manner by accelerating the rate of PrPSc generation, while depletion of plasminogen, destabilization of its structure, and interference with the PrP-plasminogen interaction hinder PrPSc propagation. Further investigation in cell culture models confirmed an increase of PrPSc formation by plasminogen. Although molecular basis of the observed activity for plasminogen remain to be addressed, our results demonstrate that plasminogen is the first cellular protein auxiliary factor proven to stimulate PrPSc propagation.
Prions are unique infectious particles that replicate in the absence of nucleic acidsCitation1 and cause fatal neurologic disorders in mammals.Citation2 In fact, the prion particle is composed of an alternatively folded form of the cellular prion protein (PrPC) encoded by the Prnp gene. The misfolded form of PrPC, referred to as PrPSc, shares the same primary structure with PrPC,Citation3 but exhibits distinctively different biochemical and biophysical properties.Citation4,Citation5 Moreover, animals lacking expression of the Prnp gene are resistant to prion infection,Citation6 suggesting that PrPC serves as a precursor for PrPSc.
The essence of prion biosynthesis is based on the protein-only hypothesis that postulates self-perpetuating replication of the prion protein.Citation1 Although the exact replication process remains elusive, the template-assisted conversion model proposes an idea that PrPSc serves as a template to convert α-helices of PrPC into the β-sheets of PrPSc during prion replication.Citation7 According to many lines of evidence, there exists an unidentified auxiliary factor, designated protein X, which favorably interacts with PrPC to produce a thermodynamically stable intermediate conformation called PrP*.Citation8 By introducing PrPSc to a host, dimers will readily form between PrP* and PrPSc. This interaction induces PrP* to take on the conformation of PrPSc and results in a complex consisting of the template and the newly formed PrPSc molecules. Once the dimer disassociates protein X and the two PrPSc molecules are released and allowed to continue replicating in an exponential fashion. Recent observations that infectious material can be generated in vitro using recombinant PrP have authenticated the protein-only hypothesis.Citation9,Citation10 However, the infectivity of these in vitro generated PrPSc products is lower than that of brain-derived PrPSc, leading to the possibility that insufficient levels of the unidentified auxiliary factor are the limiting factor for these in vitro assays.
To date, non-mammalian chaperone proteins, sulfated glycans and certain polyanionic macromolecules, such as RNA, have shown to increase the level of PrPSc in several different in vitro assays.Citation11–Citation14 However, defining these molecules as cellular auxiliary factors that promote the conversion of PrPC to PrPSc has been prevented for several reasons. First, overexpression of yeast heat shock protein Hsp 104 in transgenic mice does not modulate the incubation time of disease and PrPSc accumulation upon prion inoculation.Citation15 Although mammalian Hsp 70 is upregulated in humans and animals with prion diseases,Citation16,Citation17 it participates in downregulation, but not upregulation, of PrPSc accumulation.Citation18 Second, the effect of sulfated glycans on PrPSc formation has been inconsistentCitation11–Citation13 and certain sulfated glycans inhibit PrPSc propagation in animals and cultured cells.Citation19–Citation21 Lastly, although RNA is able to increase PrPSc propagation and induce the formation of PrPSc de novo from purified PrPC in the absence of a PrPSc seed,Citation22 its specificity is in question. Thus, an auxiliary factor that positively assists PrPSc replication and is composed of a mammalian cellular protein remains to be identified.
Our approach to identify an auxiliary factor relies on the idea that the auxiliary factor interacts with PrPC as proposed in the protein X hypothesis.Citation7 Therefore, we considered PrP ligands as the best candidates for this unidentified factor. By screening a phage display cDNA expression library from ScN2a cells, we identified kringle domains of plasminogen that interact with recombinant PrP folded in an α-helical conformation (α-PrP).Citation23 In vitro binding assays showed that interaction between plasminogen and α-PrP that represents the conformational state of PrPC was enhanced by the introduction of a dominant negative mutation and the presence of the basic N-terminal sequences in PrP.Citation23 Interaction of PrPC and plasminogen was further confirmed by the ability of plasminogen and its kringle domains to readily interact with α-PrP.Citation24–Citation29 However, despite greater interaction with α-PrP, plasminogen also interacted with PrP in β-sheet conformations.Citation23 Prior to our study, it was shown that PrPSc was immunoprecipitated with beads linked to plasminogen, its first three kringle domains [K(1+2+3)], and more recently the repeating YRG motif found within the plasminogen kringle domains.Citation30–Citation33 However, the ability of plasminogen to bind to PrPSc was dependent on conditions of the lipid rafts and plasminogen was actually associated with PrPC in the intact lipid rafts.Citation34
To determine the functional relevance of this interaction for PrPSc replication, we explored whether plasminogen enhances PrPSc propagation using cell culture models and the in vitro PrP conversion assay, termed protein misfolding cyclic amplification (PMCA).Citation35 The addition of plasminogen in PMCA resulted in the generation of significantly more PrPSc in a concentration-dependent manner (), suggesting that plasminogen stimulates the conversion of PrPC to PrPSc. Indeed, our kinetic studies showed that plasminogen accelerates the rate of PrPSc generation during the early stages of the in vitro PrP conversion reaction (reviewed in ref. Citation35). In contrast, the addition of plasminogen in PMCA lacking either PrPC or PrPSc failed to generate PrPSc ( and C). These results suggest that both PrPC and PrPSc are required for PrPSc replication stimulated by plasminogen and that plasminogen facilitates neither spontaneous PrP conversion nor PrPSc augmentation through aggregating pre-existing PrPSc. Incubation of plasminogen with pre-formed PrPSc followed by treatments with proteinase K failed to either increase or decrease the PrPSc level compared to controls ( and E), suggesting that plasminogen is not involved in stabilization of PrPSc, enhancement of PrPSc resistance to protease or promotion of PrPSc binding to the membrane for western blotting. The activity to stimulate PrP conversion was a specific property of plasminogen and was not shared with other proteins such as a known PrP ligand and proteins abundantly found in the serum or at the extracellular matrices where plasminogen is present (reviewed in ref. Citation35). In addition, we found that the ability of plasminogen to assist in PrPSc propagation is preserved in its kringle domains (reviewed in ref. Citation35). Furthermore, the activity associated with plasminogen under cell-free conditions was reproduced in cell culture models. Plasminogen and its kringle domains increased PrPSc propagation in cultured cells chronically infected with mouse-adapted scrapie or chronic wasting disease prions (). This suggests that the activity of plasminogen in PrPSc replication has biological relevance.
Corresponding to the results that plasminogen positively assists in PrPSc replication by stimulating conversion of PrPC to PrPSc, depletion of plasminogen from the PMCA reaction by using brain material derived from plasminogen-deficient mice restricted PrPSc replication to the basal level (). Supplementation with plasminogen for PMCA using plasminogen-deficient brain homogenate restored PrPSc propagation to levels equivalent to that of control PMCA in which only brain material of their non-genetically altered littermates was used (). Furthermore, structural destabilization of plasminogen affected the activity of plasminogen that enhances PrPSc propagation. Because intact disulfide bonds are critical in maintaining structural integrity and the binding activity of plasminogen, we conducted PMCA supplemented with either structurally intact or modified plasminogen to investigate the functionality of plasminogen. The result showed that plasminogen pre-treated sequentially with chemical agents that disrupt disulfide bonds and modify free sulfhydryl groups failed to stimulate PrPSc propagation (reviewed in ref. Citation35). In addition, we showed that interference with the plasminogen-PrP interaction using L-lysine abolished the plasminogen-mediated stimulation of PrPSc propagation in PMCA (reviewed in ref. Citation35). Because previous observations in immunoprecipitationCitation30 and ELISA binding assaysCitation23 described that L-lysine specifically inhibited formation of the PrP-plasminogen complex, the presence of L-lysine in PMCA is considered to saturate the lysine binding motifs of kringle domains, which competitively prevents the kringle domains of plasminogen from interacting with PrP and inhibited PrP conversion. These various inhibition studies of PrPSc propagation provides confirmatory evidence that plasminogen plays an important role in PrPSc replication.
Despite our progress in understanding the role of plasminogen in PrPSc propagation, we are still unable to address mechanistic details by which plasminogen exerts its function. In fact, plasminogen shares a number of the expected characteristics of the previously proposed auxiliary factor, although there are minor but distinctive discrepancies in their properties (summarized in ). With the insight obtained from known properties of plasminogen, it is possible to postulate a few plausible mechanisms how plasminogen promotes PrPSc replication (). First, plasminogen may control conformational rearrangement of PrPC to PrP*, resembling a molecular chaperone. This scenario is identical to the protein X hypothesis, in which plasminogen replaces protein X to assist the conversion process. Second, plasminogen may promote aggregation of PrPSc already converted from PrPC. This will result in efficient formation of PrPSc multimers. Third, plasminogen may stabilize the pre-existing PrPSc aggregates so that stabilized aggregates are better protected from degradation or processing by the intrinsic clearance mechanism. This will result in increased accumulation and stability of PrPSc. In the second and third scenarios, the function of plasminogen is not involved in PrPC or its conversion process, but limited to the interaction with pre-formed PrPSc. Fourth, plasminogen may play a role as a scaffolding molecule that simply brings both PrPC and PrPSc together within a proximity. This will increase the frequency of interaction between PrPC and PrPSc for conversion. This scenario is distinguished from the other three by postulating plasminogen interaction with both isoforms of PrP. In contrast, plasminogen is assumed to interact with only PrPC or PrPSc in other cases. Although accumulating data including our recent studies provide critical pieces of evidence to envision described mechanistic insights, it is still premature to conclude the mechanism involved in plasminogen-mediated stimulation of PrPSc propagation.
It is essential to address a few additional issues of PrPSc propagation stimulated by plasminogen aside from the mechanistic details. First, it is necessary to confirm the authenticity of PrPSc generated in the aid of plasminogen as a bona fide infectious agent. In addition, it would be interesting to compare the structural signatures of PrPSc generated in our study and found the prion seeds. This study may provide a clue to establish a structure-infectivity relationship for PrPSc. Furthermore, the ability to repeat our PMCA results in a similar assay reconstituted with defined components would clarify whether plasminogen directly contributes to PrPSc propagation. Finally, the contribution of plasminogen to the species barrier by controlling the compatibility between the host PrPC and prion strains should be determined. Collectively, these studies will be useful to identify an intricate regulatory role for plasminogen during PrPSc replication and prion transmission.
Plasminogen has shown that it stimulates PrPSc propagation in the cell-free assays and cultured cells, while its role in animal models has not been obviously clarified. In opposition to the anticipated outcomes that the course of prion disease in plasminogen-deficient animals would be delayed, intracerebral prion infection of two independent mouse lines deficient in plasminogen resulted in either unchanged or accelerated disease progression when compared to the appropriate wild-type controls.Citation36,Citation37 These results do not support each other and there is no good interpretation to reconcile their incongruence. However, the discrepancy between the expectation and the actual outcomes of the previous studies can be caused by the intrinsic health problems associated with the mouse lines used for the studies,Citation38–Citation40 which raises a question for the credibility of the model system. Some clinical phenotypes of confounding health problems in plasminogen-deficient mice overlap with the typical clinical signs of prion disease in mice. Furthermore, the drastically shortened life expectancy of plasminogen-deficient mice coincides with the incubation periods of mice inoculated with prion strains used in the previous studies. Lastly, the health problems of plasminogen-deficient mice could exacerbate the progression of the course of disease in mice inoculated with prions. Thus, either unchanged or shortened incubation periods of prion-inoculated plasminogen-deficient mice may not solely reflect the effect of plasminogen deficiency related to prion replication. Alternatively, the reason for observed outcomes from the in vivo models may be associated with the presence of functionally redundant proteins for plasminogen even under its absence. Because kringle domains of plasminogen share a well-conserved structure with those of other proteins such as tissue-type plasminogen activator, hepatocyte growth factor and apolipoprotein (a),Citation41 it is possible to postulate that other proteins that contain kringle domains can functionally replace plasminogen. Therefore, an animal model that circumvents the drawbacks associated with the current models is needed to address the relevance of plasminogen in prion replication in vivo.
Based on our study that identified plasminogen as the first cellular protein cofactor for PrPSc propagation, we anticipate an intriguing opportunity to develop future diagnosis and therapeutic intervention for prion disease. Previously, the identity of a proposed auxiliary factor was obscure so most approaches either targeted PrP isoforms or were empirical.Citation42 Our study suggests that plasminogen is a novel target to interfere with PrPSc replication. Therefore, a variety of strategies that either deplete plasminogen or interfere with the formation of PrP-plasminogen complexes would work for development of a novel therapeutic intervention for prion disease. For instance, RNA interference of plasminogen expression, monoclonal anti-plasminogen antibody to block plasminogen binding to PrP, L-lysine that saturates the binding sites of plasminogen to PrP, and chemical agents that destabilize the structure of plasminogen are potential strategies for this purpose.
In conclusion, plasminogen is a PrP ligand with the ability to stimulate PrPSc propagation. Our findings are indispensable in gaining a better understanding of the underlying mechanism for PrPSc propagation, while unveiling a new therapeutic target for prion disease.
Abbreviations
| PrP | = | prion protein |
| PrPSc | = | disease-associated prion protein |
| PrPC | = | cellular prion protein |
| Prnp | = | the gene that encodes PrPC |
| protein X | = | unidentified auxiliary factor for PrP conversion |
| PrP* | = | an intermediate conformation of PrP generated from PrPC |
| K(1+2+3) | = | the first three kringle domains of plasminogen |
| α-PrP | = | PrP in an α-helical conformation |
| β-PrP | = | PrP in an β-sheet conformation |
| PMCA | = | protein misfolding cyclic amplification |
Figures and Tables
Figure 1 The role of plasminogen in PrPSc propagation. The effect of plasminogen (Plg) was assessed by PMCA using normal brain material supplemented with or without 0.5 µM human Glu-Plg (A–D) or using Plg-deficient (Plg-/-) brain material (F and G). Pre- (−) and post- (+) PMCA samples were treated with proteinase K (PK) and analyzed by western blotting. Seeds for PMCA were diluted either as indicated or 1:900 (G) −8,100 (F). (A) Stimulation of PrPSc propagation by Plg. (B) Plg-supplemented PMCA in the absence of SBH seeds. (C) Plg-supplemented PMCA in the absence of NBH. (D) Comparison of PrPSc levels in Plg-supplemented PMCA samples (during) vs. PMCA samples only incubated with Plg prior to PK digestion (after). (E) Comparison of PrPSc levels of ScN2a cell lysate after incubation with or without Plg prior to PK digestion. (F) PMCA with brain material of Plg-/- mice and genetically unaltered littermate controls (C). (G) Restoration of PMCA using Plg-/- brain material with Plg-supplementation. NBH, normal brain homogenate; SBH, sick brain homogenate; PrPKOBH, brain homogenate of PrPC-deficient mice; CL, cell lysate. Reproduced with permission from The FASEB Journal, Mays and Ryou 2010.Citation35
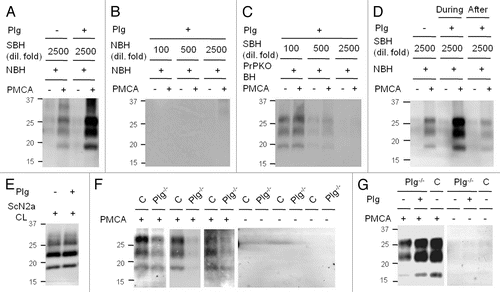
Figure 2 PrPSc propagation increased by plasminogen in prion-infected cells. (A) The levels of PrP in ScN2a cells incubated with 0–0.5 µM human Glu-plasminogen (Plg) for two days. (B) The levels of PrP in ScN2a cells incubated with 0, 0.1 and 1.0 µM Plg or the first three kringle domains of Plg [K(1+2+3)] for six days. (C) The levels of 3F4-tagged PrPC and nascent PrPSc formation in ScN2a cells transiently transfected (Tfx) with plasmids encoding the 3F4-tagged PrP gene (PrP-3F4) and with an empty vector (mock). The transfected cells were treated with 0 or 1 µM K (1+2+3) for three days. (D) The levels of PrP in Elk21+ cells incubated with 0 and 0.5 µM Plg for two days. PrP was detected by anti-PrP antibody D13 (A and B), 3F4 (C) or 6H4 (D) before (−) and after (+) PK treatment. Reproduced by permission of the The FASEB Journal, Mays and Ryou 2010.Citation35
![Figure 2 PrPSc propagation increased by plasminogen in prion-infected cells. (A) The levels of PrP in ScN2a cells incubated with 0–0.5 µM human Glu-plasminogen (Plg) for two days. (B) The levels of PrP in ScN2a cells incubated with 0, 0.1 and 1.0 µM Plg or the first three kringle domains of Plg [K(1+2+3)] for six days. (C) The levels of 3F4-tagged PrPC and nascent PrPSc formation in ScN2a cells transiently transfected (Tfx) with plasmids encoding the 3F4-tagged PrP gene (PrP-3F4) and with an empty vector (mock). The transfected cells were treated with 0 or 1 µM K (1+2+3) for three days. (D) The levels of PrP in Elk21+ cells incubated with 0 and 0.5 µM Plg for two days. PrP was detected by anti-PrP antibody D13 (A and B), 3F4 (C) or 6H4 (D) before (−) and after (+) PK treatment. Reproduced by permission of the The FASEB Journal, Mays and Ryou 2010.Citation35](/cms/asset/f7dff541-457f-4713-b345-58ccb5c01505/kprn_a_10914460_f0002.gif)
Figure 3 Plausible mechanisms for plasminogen to enhance PrPSc propagation. Plasminogen may stimulate PrPSc propagation via conformational alteration of PrPC to PrP* (i), enhancement of PrPSc aggregation (ii), stabilization of pre-exisiting PrPSc aggregates (iii) or scaffolding to gather PrPC and PrPSc together (iv).
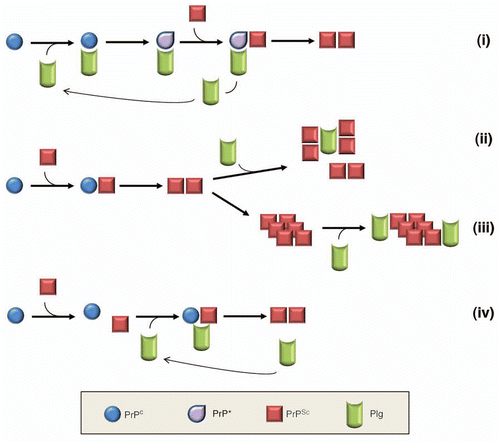
Table 1 Properties of plasminogen as an auxiliary factor for PrPSc propagation
References
- Prusiner SB. Novel proteinaceous infectious particles cause scrapie. Science 1982; 216:136 - 144
- Prusiner SB. Prions. Proc Natl Acad Sci USA 1998; 95:13363 - 13383
- Basler K, Oesch B, Scott M, Westaway D, Walchli M, Groth DF, et al. Scrapie and cellular PrP isoforms are encoded by the same chromosomal gene. Cell 1986; 46:417 - 428
- Pan KM, Baldwin M, Nguyen J, Gasset M, Serban A, Groth D, et al. Conversion of a-helices into b-sheets features in the formation of the scrapie prion proteins. Proc Natl Acad Sci USA 1993; 90:10962 - 10966
- Caughey BW, Dong A, Bhat KS, Ernst D, Hayes SF, Caughey WS. Secondary structure analysis of the scrapie-associated protein PrP 27–30 in water by infrared spectroscopy. Biochemistry 1991; 30:7672 - 7680
- Bueler H, Aguzzi A, Sailer A, Greiner RA, Autenried P, Aguet M, et al. Mice devoid of PrP are resistant to scrapie. Cell 1993; 73:1339 - 1347
- Prusiner SB. Molecular biology of prion diseases. Science 1991; 252:1515 - 1522
- Cohen FE, Prusiner SB. Pathologic conformations of prion proteins. Annu Rev Biochem 1998; 67:793 - 819
- Kim J, Cali I, Surewicz K, Kong Q, Raymond GJ, Atarashi R, et al. Mammalian prions generated from bacterially expressed prion protein in the absence of any mammalian cofactors. J Biol Chem 2010; 285:14083 - 14087
- Makarava N, Kovacs GG, Bocharova O, Savtchenko R, Alexeeva I, Budka H, et al. Recombinant prion protein induces a new transmissible prion disease in wild-type animals. Acta Neuropathol 2010; 119:177 - 187
- Deleault NR, Geoghegan JC, Nishina K, Kascsak R, Williamson RA, Supattapone S. Protease-resistant prion protein amplification reconstituted with partially purified substrates and synthetic polyanions. J Biol Chem 2005; 280:26873 - 26879
- Deleault NR, Lucassen RW, Supattapone S. RNA molecules stimulate prion protein conversion. Nature 2003; 425:717 - 720
- Wong C, Xiong LW, Horiuchi M, Raymond L, Wehrly K, Chesebro B, et al. Sulfated glycans and elevated temperature stimulate PrPSc-dependent cell-free formation of protease-resistant prion protein. EMBO J 2001; 20:377 - 386
- DebBurman SK, Raymond GJ, Caughey B, Lindquist S. Chaperone-supervised conversion of prion protein to its protease-resistant form. Proc Natl Acad Sci USA 1997; 94:13938 - 13943
- Dandoy-Dron F, Bogdanova A, Beringue V, Bailly Y, Tovey MG, Laude H, et al. Infection by ME7 prion is not modified in transgenic mice expressing the yeast chaperone Hsp104 in neurons. Neurosci Lett 2006; 405:181 - 185
- Kenward N, Hope J, Landon M, Mayer RJ. Expression of polyubiquitin and heat-shock protein 70 genes increases in the later stages of disease progression in scrapie-infected mouse brain. J Neurochem 1994; 62:1870 - 1877
- Shyu WC, Kao MC, Chou WY, Hsu YD, Soong BW. Creutzfeldt-Jakob disease: heat shock protein 70 mRNA levels in mononuclear blood cells and clinical study. J Neurol 2000; 247:929 - 934
- Fernandez-Funez P, Casas-Tinto S, Zhang Y, Gomez-Velazquez M, Morales-Garza MA, Cepeda-Nieto AC, et al. In vivo generation of neurotoxic prion protein: Role for Hsp70 in accumulation of misfolded isoforms. PLoS Genet 2009; 5:1000507
- Ehlers B, Diringer H. Dextran sulphate 500 delays and prevents mouse scrapie by impairment of agent replication in spleen. J Gen Virol 1984; 65:1325 - 1330
- Kimberlin RH, Walker CA. Suppression of scrapie infection in mice by heteropolyanion 23, dextran sulfate and some other polyanions. Antimicrob Agents Chemother 1986; 30:409 - 413
- Caughey B, Raymond GJ. Sulfated polyanion inhibition of scrapie-associated PrP accumulation in cultured cells. J Virol 1993; 67:643 - 650
- Deleault NR, Harris BT, Rees JR, Supattapone S. Formation of native prions from minimal components in vitro. Proc Natl Acad Sci USA 2007; 104:9741 - 9746
- Ryou C, Prusiner SB, Legname G. Cooperative binding of dominant-negative prion protein to kringle domains. J Mol Biol 2003; 329:323 - 333
- Cuccioloni M, Amici M, Eleuteri AM, Biagetti M, Barocci S, Angeletti M. Binding of recombinant PrPc to human plasminogen: Kinetic and thermodynamic study using a resonant mirror biosensor. Proteins 2005; 58:728 - 734
- Ellis V, Daniels M, Misra R, Brown DR. Plasminogen activation is stimulated by prion protein and regulated in a copper-dependent manner. Biochemistry 2002; 41:6891 - 6896
- Epple G, Langfeld K, Baier M, Holzhütter HG, Schleuning WD, Köttgen E, et al. Both lysine-clusters of the NH2-terminal prion-protein fragment PrP23–110 are essential for t-PA mediated plasminogen activation. Thromb Haemost 2004; 91:465 - 471
- Epple G, Schleuning WD, Kettelgerdes G, Kottgen E, Geßner R, Praus M. Prion protein stimulates tissue-type plasminogen activator-mediated plasmin generation via a lysine-binding site on kringle 2. J Thromb Haemost 2004; 2:962 - 968
- Kornblatt JA, Marchal S, Razaei H, Balny C. Characterization of a complex formed between human plasminogen and recombinant sheep prion: pressure and thermal sensitivity of complex formation. Cell Mol Biol (Noisy-le-grand) 2004; 50:387 - 396
- Praus M, Kettelgerdes G, Baier M, Holzhütter HG, Jungblut PR, Maissen M, et al. Stimulation of plasminogen activation by recombinant cellular prion protein is conserved in the NH2-terminal fragment PrP23–110. Thromb Haemost 2003; 89:812 - 819
- Fischer MB, Roeckl C, Parizek P, Schwarz HP, Aguzzi A. Binding of disease-associated prion protein to plasminogen. Nature 2000; 408:479 - 483
- Maissen M, Roeckl C, Glatzel M, Goldmann W, Aguzzi A. Plasminogen binds to disease-associated prion protein of multiple species. Lancet 2001; 357:2026 - 2028
- Hatcher K, Zheng J, Chen SG. Cryptic peptides of the kringle domains preferentially bind to disease-associated prion protein. J Alz Dis 2009; 16:421 - 431
- Negredo C, Monks E, Sweeney T. A novel real-time ultrasonic method for prion protein detection using plasminogen as a capture molecule. BMC Biotechnol 2007; 7:43
- Shaked Y, Engelstein R, Gabizon R. The binding of prion proteins to serum components is affected by detergent extraction conditions. J Neurochem 2002; 82:1 - 5
- Mays CE, Ryou C. Plasminogen stimulates propagation of protease-resistant prion protein in vitro. FASEB J 2010; 24:5102 - 5112
- Salmona M, Capobianco R, Colombo L, De Luigi A, Rossi G, Mangieri M, et al. Role of plasminogen in propagation of scrapie. J Virol 2005; 79:11225 - 11230
- Xanthopoulos K, Paspaltsis I, Apostolidou V, Petrakis S, Siao CJ, Kalpatsanidis A, et al. Tissue plasminogen activator in brain tissues infected with transmissible spongiform encephalopathies. Neurobiol Dis 2005; 20:519 - 527
- Bugge TH, Flick MJ, Daugherty CC, Degen JL. Plasminogen deficiency causes severe thrombosis but is compatible with development and reproduction. Genes Dev 1995; 9:794 - 807
- Ploplis VA, Carmeliet P, Vazirzadeh S, Van Vlaenderen I, Moons L, Plow EF, et al. Effects of disruption of the plasminogen gene on thrombosis, growth and health in mice. Circulation 1995; 92:2585 - 2593
- Degen JL, Drew AF, Palumbo JS, Kombrinck KW, Bezerra JA, Danton MJS, et al. Genetic manipulation of fibrinogen and fibrinolysis in mice. Ann NY Acad Sci 2001; 936:276 - 290
- Castellino FJ, McCance SG. The kringle domains of human plasminogen. Ciba Found Symp 1997; 212:46 - 60
- Trevitt CR, Collinge J. A systematic review of prion therapeutics in experimental models. Brain 2006; 129:2241 - 2265