Abstract
Prion diseases are fatal transmissible neurodegenerative disorders. In the pathogenesis of the disease, the cellular prion protein (PrPC) is required for replication of abnormal prion (PrPSc), which results in accumulation of PrPSc. Although there have been extensive studies using Prnp knockout systems, the normal function of PrPC remains ambiguous. Compared with conventional germline knockout technologies and transient naked siRNA-dependent knockdown systems, newly constructed durable chained-miRNA could provide a cell culture model that is closer to the disease status and easier to achieve with no detrimental sequelae. The selective silencing of a target gene by RNA interference (RNAi) is a powerful approach to investigate the unknown function of genes in vitro and in vivo. To reduce PrPC expression, a novel dual targeting-microRNA (miRdual) was constructed. The miRdual, which targets N- and C- termini of Prnp simultaneously, more effectively suppressed PrPC expression compared with conventional single site targeting. Furthermore, to investigate the cellular change following PrPC depletion, gene expression analysis of PrPC interacting and/or associating genes and several assays including proliferation, viability and apoptosis were performed. The transcripts 670460F02Rik and Plk3, Ppp2r2b and Csnk2a1 increase in abundance and are reported to be involved in cell proliferation and mitochondrial-mediated apoptosis. Dual-targeting RNAi with miRdual against Prnp will be useful for analyzing the physiological function of PrPC in neuronal cell lines and may provide a potential therapeutic intervention for prion diseases in the future.
Introduction
Prion diseases are group of fatal transmissible neurodegenerative disorders characterized by a long incubation period and broad neuropathology including spongiform encephalopathy, gliosis and neuronal loss.Citation1 They are classified into sporadic, inherited and acquired forms, which can spread within and between mammalian species.Citation2 The central feature of prion diseases is the conversion of a host-encoded prion protein (PrPC) into a disease-associated isoform (PrPSc), which shares same amino acid sequence with PrPC but is posttranslationally converted to the infectious form, accumulating primarily in the central nerve system.Citation3 PrPC is implicated in prion pathogenesis as it is required for propagation and development of prion pathology.Citation4 Although loss of PrPC function or deposition of PrPSc is not sufficient to cause the disease, the process of conversion into PrPSc may alter signal transduction, thereby giving a toxic dominant function.Citation5–Citation7
There have been extensive efforts to develop prion disease models in vivo, especially Prnp knockout models for defining the physiological role of PrPC.Citation8,Citation9 These transgenic animal models are, however, expensive and time consuming.Citation10,Citation11 To overcome these problems, cell culture models of prion diseases have been used for screening and elucidating the mechanism of action of anti-prion agents and to analyze biological properties of PrPC at the molecular and cellular levels.Citation7,Citation11,Citation12 Elucidating the cellular function of PrPC may help in deciphering mechanisms of prion pathogenesis and in devising therapeutic strategies.Citation5,Citation13
Mature PrPC translocates to the outer leaflet of the plasma membrane in proximity to raft-associated signaling molecules. It traffics in and out of lipid rafts and is involved in a diversity of molecular and physiological functions.Citation14–Citation16 These properties of PrPC suggest the possibility that several changes in Prnp knockout cell culture models may be caused by an altered expression of interlinked genes with different cellular functions. Prnp knockouts could reflect not only the missing function but also the compensation for that missing function, as gene deletions in individual cells or tissues are often accompanied by compensatory changes in gene expression patterns.Citation17,Citation18
RNA interference (RNAi) refers to the use of 21–23 nucleotide short interfering RNAs (siRNAs) mediating post-transcriptional degradation or translational repression of homologous gene transcripts. Stable knockdowns can be obtained by constitutive expression of the siRNA from the host chromosome.Citation19,Citation20 RNAi has been accepted as the most powerful reverse-genetics approach in mammalian cells, however, there are significant technical limitations including identifying efficient methods to design and deliver siRNA.Citation21 Unlike other approaches, such as traditional gene targeting by homologous recombination, antisense vectors and catalytic RNA or DNA molecules, RNAi is an endogenous natural pathway and allows cross-species application.Citation12 RNAi, which acutely decreases target expression, also has the advantage of avoiding compensatory or mechanistic adaptations.Citation18
The present study describes the development of a prion knockdown cell culture model by introducing artificial microRNA (miR) targeting Prnp into the mouse neuroblastoma cell line (N2a) as well as RK13 cells expressing mouse PrPC. The established Prnp knock-down N2a cells were characterized by investigating the expression profiles of the genes known as PrPC interacting and/or associating molecules, proliferation, viability and apoptotic resistance.
Results
Establishment of stable Prnp knockdown cells.
Knockdown of a gene using exogenously introduced siRNA is always transient due to a cell division and/or degradation of siRNA molecules.Citation22 To develop stable Prnp knockdown cells, small hairpin structural artificial miRs were designed (). The direction and sequence of the artificial miRs were confirmed by sequence analysis, and N2a cells were transfected with the either miR1, miR2, miRdual or miRscr constructs. The transient effects of each miR on prion protein expression were assessed by western blot analysis. The miRdual most effectively knocked down the expression of PrPC by 75 ± 2% compared with wild-type N2a cells (p < 0.01), whereas PrPC level in N2a cells transfected with miR1, miR2 and miRscr was decreased by 17 ± 7%, 24 ± 9% (p < 0.05) and no knockdown effect, respectively ( and C). To establish stable Prnp knockdown cells, the N2a cells transfected with miRdual or miRscr were selected with Blasticidin S for a month and are referred to here as N2amiRdual and N2amiRscr, respectively. The single clones were further selected by limiting dilution and clonally expanded cells were analyzed by genomic PCR, qPCR and western blot to confirm stable transformation with miR as well as transcription and expression of Prnp. Association of the miR plasmid with host chromosomal sequences was assessed by PCR of genomic DNA. N2amiRdual tested positive for the 550 bp amplicon, while N2amiRscr yielded 412 bp amplicon. No amplicon occurred with genomic DNA from wild-type N2a cells (). To quantify the effect of miR on Prnp transcription, qPCR was used and transcription input of each sample was normalized using qPCR of Gapdh. The Prnp transcript was decreased by 87 ± 1% in N2amiRdual (p < 0.01), while it was 50 ± 15% lower in N2amiRscr compared with wild-type N2a (p < 0.05) (). PrPC expression was quantified by western blot. Band intensity analysis of western blot indicated that the resultant protein expression of PrPC was decreased in N2amiRdual by 96 ± 1% and 94 ± 2% in two different cell clones compared with wild-type (p < 0.01). N2amiRscr showed a reduced expression of PrPC by 27 ± 4% (p < 0.05) ().
Differential gene expression in stably transformed N2amiRdual cells.
We next analyzed gene expression changes in response to Prnp knockdown. These studies were undertaken in N2a cells and N2amiRdual derivative and corresponded to the quantified expression of several target genes thought to interact and/or associate with PrPC (). For all targets, transcript inputs were normalized using Gapdh and standard curves were generated using serially diluted PCR products of each specific gene. Relative expression levels were expressed as percent compared to wild-type N2a cells. Of the seven targets selected, 670460F02Rik, Csnk2a1, Plk3 and Ppp2r2b increased in abundance (220 ± 33%, 230 ± 52%, 183 ± 10% and 519 ± 5%, respectively) in N2amiRdual cells compared with wild-type N2a cells (p < 0.01) (). The abundance of these transcripts also increased, but to a less degree, in N2amiRscr (121 ± 6%, 132 ± 11%, 147 ± 16% and 350 ± 21% respectively). The trend in expression patterns for all target genes was negatively correlated with the amount of PrPC in N2amiRdual and N2amiRscr cells ( and C). For Mpg, the mRNA expression was increased both in N2amiRdual and N2amiRscr by 195 ± 21% and 184 ± 24%, respectively (p < 0.05) (). As expected, the Cdr34 and Gfap transcripts as controls were not detectable until 40 cycles of qPCR reaction in both N2amiRs and wild-type N2a cells.
Alteration in cellular characteristics following PrPC knock-down.
After establishment of the prion knockdown cell line, N2amiRdual, proliferation, viability and mitochondrial-mediated apoptosis were examined. In proliferation assays, the N2amiRdual showed decreased proliferation, to 88 ± 4%, in mitochondrial dehydrogenase activity tests compared with wild-type (p < 0.05). No significant difference was observed in lactate dehydrogenase tests for cell viability (). Based on the observed morphology of the cell body and neuritis, no differences were observed in N2amiRdual and N2amiRscr compared with wild-type. Both cell types expressed the GFP reporter molecules, however, the signal was stronger in miRscr cassette than in miRdual (). GFP signals derived from miR expression cassette were attenuated in N2amiRdual cells, likely due to increased processing of the miRdual.
To measure mitochondrial-mediated apoptosis induced by serum withdrawal, the expression level of mitochondria fission related protein, dynamin related protein 1 (Drp1), was quantified. Drp1 expression was increased in N2amiRdual by 112 ± 4% 24 h following apoptosis induction when compared to the level before apoptosis induction (p < 0.05). There was, however, no significant difference in the Drp1 expression level between N2amiRscr and N2a wild-type, regardless of growth condition such as serum deprivation (). The level of pro-apoptotic Cytochrome C (Cyt c) in cytosol increased 215 ± 2% in N2amiRdual cells during serum deprivation compared to the level before serum deprivation (p < 0.05). Again there was no difference between N2amiRscr and wild-type N2a cells even though cells had undergone apoptotic stimulation (). During apoptotic stimulation, morphological changes between each cell lines were observed by light microscopy. Neurite retraction was induced and proliferation inhibited in N2amiRdual cell 24 h post serum deprivation, while N2amiRscr and wild-type N2a cells maintained their structural integrity. The caspase 3 activity in N2amiRdual cells was increased from 1.6 pmol pNA liberated/hour at 0 h to 18.8 pmol pNA liberated/hour at 48 h post serum deprivation (p < 0.01), whereas there were no significant differences in N2amiRscr and wild-type N2a cells (). The proportion of apoptotic cells bound to Annexin V was increased by 212 ± 4% in N2amiRdual at 24 h post apoptosis induction when compared to the percentage of dying cells before apoptosis induction (p < 0.05). Most N2amiRscr and wild-type N2a cells were resistant to apoptotic stimulation regardless of growth environment ().
Lentiviral transduction of RK13 expressing mouse PrPC with miRs.
To confirm the reduction of PrPC levels by the dual knockdown method, a stable clone of rabbit kidney epithelial cells (RK13),Citation23 expressing both mouse PrPC (RK13moPrnp) and GFP was transduced with lentivirus containing either the miRdual or miRscr expression cassettes. The cells were harvested at 70% and 100% confluence and the relative abundance of PrPC analyzed by western blot (). In these analyses quantities of PrPC and GFP were normalized to the sample with either the miRdual or miRscr at 70% cell confluency. PrPC level was decreased in RK13moPrnp cells with miRdual (46 ± 8% and 58 ± 5%), while no significant difference in PrPC level was observed in RK13moPrnp cells with miRscr (p < 0.05) (). Besides assessing protein loading with an actin probe, the GFP also encoded within the bigenic plasmidCitation24 was evaluated by use of an anti-GFP antibody. No diminution was seen when using the “dual” lentivirus, speaking to specificity, and in fact GFP protein levels were elevated by ∼30% both in RK13moPrnp-miRdual (136 ± 4% and 139 ± 7%) and in RK13moPrnp-miRscr cells (127 ± 6% and 129 ± 4%). This effect cannot represent alleviation of a transcription interference effect from Prnp cassette within the bigenic pBUD vector (which has tandem expression cassettes), as it is seen with viruses with different effects upon PrP expression.
Discussion
The knockdown of PrPC in N2a cells using a miR-based RNAi system provides a novel cell culture model for studying prion biology. The artificial miRs were designed to target nucleotide residues 36–56 and 668–688 of mouse PrPC ORF region, respectively. The miRs were constructed to be expressed as a cluster in a long primary transcript driven by RNA polymerase II. This dual targeting strategy enhanced knockdown efficacy more than 3-fold compared to single site targeting method. It was reported that a single miR can bind to and regulates many different mRNA target, conversely, several different miRs can pair with and cooperatively control a single mRNA target.Citation25,Citation26 Downregulation of Prnp was also observed in N2a transfected with miRscr, an unexpected result as the scrambled sequence does not target any known vertebrate gene. To better define this targeting effect of miRscr to Prnp, we examined RK13moPrnp cells, which were lentiviral-transduced with miRdual or miRscr, respectively. In the viral-tranduced cells with miRdual, the expression levels of PrPC varied inversely with the amount of GFP. These results suggest that, rather than specific targeting by miRscr, the reduced PrPC expression in N2amiRscr is likely due to exogenous factors through gene transfection, such as genomic insertion of foreign genes that may disrupt cellular processes.Citation27–Citation29
An exploration of cellular interactors is one of the way to disclose unknown function of protein of interest.Citation14 Transcript abundances of PrPC interacting and/or associating proteins following stable Prnp knockdown can provide valuable information concerned with normal cellular function of PrPC. Therefore, we selected seven target genes from previously reported microarray and protein microarray dataCitation30–Citation33 and transcript abundance was investigated by qPCR. Of the seven genes examined in this study, the abundance of four PrPC interactors increased following Prnp downregulation. One of the upregulated genes in N2amiRdual is 6720460F02Rik gene, also known as CATS (computer-annotated as FAM64A), the Homo sapiens family with sequence similarity 64, member A in GenBank (NM_144526). Its transcript is abundantly expressed in proliferative state brain tissues, glioma and primitive neuroectodermal tumors, however, the precise cellular function of the protein is unknown.Citation30 FAM64A was shown to interact with PrPC through protein microarray and coimmunoprecipitation with recombinant human PrPC spanning amino acid residues 23–231. In addition, FAM64A is located predominantly in the nucleus, where it colocalizes with the recombinant human PrPC.Citation30 This suggests that FAM64A/CATS/6720460F02Rik might have a role in control of cell proliferation consistent with the decreased cell proliferation observed in our studies.Citation34 The increased transcription level could be a compensatory reaction to the loss of PrPC function associated with cell proliferation.
Plk3 is a member of the polo family of serine/threonine kinases and is also as a PrP interacting partner.Citation30 It localizes to the nucleolus and is involved in regulation of the G1/S phase transition.Citation35,Citation36 The control of cell proliferation by PrPC may alter the expression of cell cycle-related genes, such as cyclin D1, Esp8 and CD44, in a cell type-specific way.Citation37 The increased level of Plk3 in N2amiRdual cells may be a compensatory response to a PrPC-dependent cell proliferation event. Moreover, the overexpression of Plk3 may mediate inhibition of proliferation as a result of the loss of the anti-apoptotic function of PrPC.Citation36,Citation37 Further analysis into the mechanism of PrPC function associated with Plk3 will be required.
Ppp2r2b encodes the neuron-specific regulatory beta subunit of the protein phosphatase 2A (PP2A) holoenzyme and the mutations in Ppp2r2b are responsible for the neurodegenerative disorder, spinocerebellar ataxia 12. Increased expression of Ppp2r2b reported to induce mitochondrial fragmentation through stimulating mitochondrial fission protein such as Drp1, which can lead to neuronal apoptosis.Citation38,Citation39 Our observation of increased expression of Ppp2r2b following PrPC knockdown was not consistent with previous study that showed the increased Ppp2r2b in a PrPC overexpressing HEK293 cell line.Citation31 It is conceivable that dysregulation of Ppp2r2b via abnormal expression of PrPC might be a critical cause of cell death secondary to mitochondrial dysfunction. Mitochondrial fragmentation by overexpression of Ppp2r2b occurs upstream of apoptosis and was sufficient for hippocampal neuron death.Citation39 It is consistent that transfection of PrP knockout cells with a Prnp expression vector prevented typical apoptotic changes.Citation37
Casein kinase 2 (CK2) participates in the regulation of diverse cellular processes. It is a messenger-independent protein serine/threonine kinase and especially abundant in the brain. It is localized to the outer plasma membrane where PrPC also resides.Citation40 The catalytic α subunit of CK2 (CK2α), encoded by the Csnk2a1 gene, is reported to bind to cytosolic non-glycosylated PrPC.Citation41 CK2α also binds to PP2A in mitogen-starved cells in vitro and overexpression of CK2α results in suppression of cell growth.Citation42 These reports are consistent with the result of decreased proliferation in N2amiRdual cells that may cause compensatory increases of Ppp2r2b and Csnk2a1 following decreased expression level of PrPC.
The Mpg gene encodes the base-excision repair enzyme, 3-methyladenine DNA glycosylase and has been reported as a PrP interacting protein.Citation30 Mpg plays a vital role in preserving cerebellar development and protecting mature neurons from insults by environmental genotoxins.Citation43,Citation44 The upregulation of Mpg both in N2amiRdual and N2amiRscr is curious, but may be a response to the expression of double-stranded miR that has internal loops.
There was no difference observed in viability of the N2a lines but proliferation was decreased in N2amiRdual compared with N2amiRscr and wild-type cells. This is likely due to the downregulation of PrPC rather than other exogenous factors. It has been shown that the expression level of PrPC was positively correlated with the cell proliferation.Citation37,Citation45 In agreement with other studies of prion knockout model,Citation46–Citation48 newly constructed prion knockdown N2amiRdual cells were more vulnerable to apoptotic stimulation than N2amiRscr and wild-type N2a. Inhibition of Drp1 activity during apoptosis in mammalian cells inhibits mitochondrial fragmentation, Cyt c release and the rate of cell death.Citation39,Citation49,Citation50 Altering the mitochondrial fusion/fission balance towards fission by Drp1 could lead to cell death by promoting mitochondrial release of pro-apoptotic proteins including Cyt c.Citation39 The release of Cyt c from mitochondrial inter-membrane space to the cytosol has reported to activate the caspase family,Citation51,Citation52 an important event for the downstream activation of apoptosis cascade.Citation49,Citation53 This phenomenon is supported by Cyt c knockout cell lines study showing reduced caspase 3 activation and resistance to various apoptotic stimuli.Citation54
In conclusion, the stable N2amiRdual cells will be a useful tool for elucidating the physiological function of PrPC and neuropathogenesis of prion disease. Moreover, since miR is known as more suitable approach for achieving RNAi in mouse brain, in terms of toxicity,Citation55 it is expected that the dual targeting artificial miR-based RNAi used in this study will provide a promising therapeutic intervention for prion diseases and other neurodegenerative disorders.
Materials and Methods
Cell culture.
N2a and RK13moPrnp cells were maintained in Dulbecco's modified Eagle's Medium (DMEM) with high glucose (4.5 g/l) and 2 mM glutamine (Gibco®, Invitrogen, #11995), supplemented with 10% fetal bovine serum, penicillin and streptomycin. Cells were cultured at 37°C in a 5% CO2 incubator.
Designing artificial miRNAs.
miRs were designed to knockdown the endogenous Mus musculus Prnp in N2a cells based on previous report in reference Citation56. The miR design algorithm, provided by the Invitrogen web-site,Citation57 was used to select target sequences. The basic local alignment search tool (BLAST) was used with the mouse genome database to identify unique regions in the murine Prnp open reading frame (ORF, NM_011170). Two regions were chosen to target; the N-(miR1) and C-termini (miR2) of the Prnp ORF. miR1 and miR2 bind at 191 to 211 and 823 to 843 of Prnp ORF region, respectively. Each single-stranded DNA (ssDNA) was designed to contain the antisense sequence derived from target sites, followed by 19 nucleotides to form the terminal loop and sense target sequence with 2 nucleotides removed to create an internal loop ().
Construction and expression of miRNAs.
The miRs were synthesized commercially (Invitrogen) and annealed for directional cloning. The synthesized top- and bottom-strand DNAs () were annealed and ligated into pcDNA6.2-GW/EmGFP-miR (Invitrogen, #K4936-00) expression vector. The ligation mixture was transformed into TOP10 competent E. coli. To confirm the orientation and DNA sequence of the miRs, each construct was sequenced using an automated DNA Sequencer (ABI PRISM 377 L). To express miRs as one primary transcript, miR2 cassette were digested and ligated into miR1 construct. The linked miRs (miRdual) was transformed and analyzed as described above. A miR cassette containing a scramble sequence (miRscr) (Invitrogen, #K4936-00), which can be processed into mature miR but is not predicted to target any known vertebrate gene, was used as a negative control ().
Transfection of N2a cells with miRNAs.
The wild-type N2a cell line was transfected with the expression construct using the FuGENE6 transfection reagent (Roche, #11814443001). At 24 h post transfection, the cells were lysed with RIPA lysis buffer (1% Triton X-100, 1% sodium deoxycholate, 150 mM NaCl, 50 mM Tris-HCl, pH 7.4, 0.1% SDS, 1 mM EDTA) containing a protease inhibitor cocktail (Amresco, #M222) and the efficiency of downregulation of Prnp was measured by western blot. Through drug selection with Blasticidin S (Invitrogen, #R21001) and limiting dilution, clonal expanded cells expressing miRdual (N2amiRdual) or miRscr (N2amiRscr) were obtained and analyzed by expression of green fluorescent protein (GFP) reporter gene, genomic PCR, quantitative PCR (qPCR) and western blot.
Transfection of RK13 cells with mouse Prnp.
RK13 cells were transfected with a derivative of the bigenic pBud mammalian vector expressing both GFP and a wt mouse Prnp coding region. This procedure used lipofectamine2000 (Invitrogen, #11668). Cells stably expressing mouse PrPC (RK13moPrnp) were selected with Zeocin (Invitrogen, #R250) and screened by western blot.
Production of lentivirus and infection of RK13moPrnp.
The miR expression cassettes including GFP were cloned into pLenti6/V5-DEST lentiviral vectors (Invitrogen, #K4937) by Gateway site-specific recombination. The constructed lentiviral vectors (3 µg) were co-transfected with a third-generation lentivirus packaging vector (9 µg, Invitrogen, # K4970) into HEK293FT cells using Lipofectamine 2000 reagent (Invitrogen, #11668). The viral supernatant was collected 48 h after co-transfection and concentrated by ultracentrifugation for 2 h at 153,725 g and 4°C. The titer of lentiviruses was determined using HT1080 cells and RK13moPrnp cells were infected with the virus particles at an MOI of 10. After Blasticidin S selection for two weeks, cells were analyzed for knockdown of PrPC by western blot.
Genomic PCR.
Genomic DNA of clonal expanded cells was isolated using a genomic DNA purification kit (Promega, #A11120). Primer pairs were designed to amplify specific miR cassettes (). The PCR parameters consisted of an initial denaturation step at 95°C for 5 min followed by 40 cycles of denaturation at 95°C for 30 sec, annealing at 60°C for 30 sec and extension at 72°C for 1 min and a final extension at 72°C for 15 min. PCR products were analyzed by electrophoresis on 1.0% agarose gel.
Western blot.
For western blot analysis, 5 µg of protein from each cell lysate was prepared and fractionated on a 12% Tris-glycine gel and transferred to a 0.2 micron nitrocellulose membrane using a dry blotting system (Invitrogen, #IB1001). The membrane was blocked with 5% nonfat dry milk in TritonX-Tris buffered saline (TTBS: 0.1% TritonX-100 in 100 mM Tris-HCl, pH 7.5, 0.9% NaCl) and then probed with anti-PrP antibody, 3F10,Citation58 anti-GFP antibody (AbCAM, #ab290) or anti-dynamin-related protein 1 (Drp1) antibody (BD Transduction Laboratories, #611112) at 4°C for overnight. An anti-mouse IgG conjugated to horseradish peroxidase (Santa Cruz Biotechnology, #SC-2005) was used as the secondary antibody. The bound antibodies were visualized by chemiluminescence (Amersham, #RPN2106) and protein levels were measured by scanning and evaluating density (Multi Gauge v2.3 software, Fujifilm). The membrane was stripped in stripping solution (100 mM Tris-HCl, pH 7.5, 0.9% NaCl, 7 µl/ml β-mercaptoethanol, 2% SDS) and then blocked with 5% nonfat dry milk in TTBS and probed with anti-mouse glyceraldehydes-3-phosphate dehydrogenase antibody (GAPDH, Santa Cruz Biotechnology, #SC-32233) or anti-mouse β-actin antibody (AbCAM, #ab20272) as a loading control. Protein levels were visualized as described above.
Quantitative PCR.
To quanify transcript abundance in N2amiRdual cells, genes reported to interact and/or associate with PrPC were selectedCitation30–Citation33 and their abundance was determined by qPCR (). Total RNA from clonal expanded cells was extracted using Trizol reagent (Invitrogen, #15596-018) and single stranded cDNA was synthesized using SuperScript™ III First-Strand Synthesis System (Invitrogen, #18080-051) with Oligo dT following the manufacturer's instructions. ORFs corresponding to each target gene were amplified by PCR using specific primers () and each PCR product was serially diluted and used to generate standard curves. The primers for qPCR were designed by D-LUX™ Designer (Invitrogen) (). Thermal cycling for the ABI PRISM 7300 (Applied Biosystems) was performed as follows: 2 min at 50°C for uracil N-glycosylase (UDG) incubation which removes uracil-containing products from previous reactions, UDG inactivation and DNA polymerase activation for 2 min at 95°C, followed by 40 cycles of 15 sec at 95°C for denaturation and 30 sec at 60°C for hybridization and elongation.
Proliferation and viability assay.
The established N2amiRdual, N2amiRscr and wild-type cell lines were cultured in 96-well plates with 3 × 104 cells in 100 µl of DMEM culture medium per each triplicate well. Activity of cellular mitochondrial and lactate dehydrogenase were determined using commercial kits (Biovision, #302-500, #313-500) to measure proliferation and cells viability respectively. After 48 h incubation, the tetrazolium salt (WST) was added to each well and the amount of formazan production quantified using a microtiter plate reader at 450 nm. Non-viable controls were prepared by treating cells with 1% TritonX-100.
Cytochrome C assay.
The cytosolic and mitochondrial fractions from each cell line were separated using a mitochondria isolation kit (Pierce, #89874) following manufacturer's instruction. Total protein concentration of the cytosolic fractions were adjusted using BCA protein assay (Pierce, #23227) and the amount of Cyt c in each fraction were analyzed by sandwich Enzyme Linked-Immuno-Sorbent Assay (Invitrogen, #KHO1051).
Caspase 3 assay.
Caspase 3 activity in cells induced to apoptosis was measured by CaspACE Assay System (Promega, #G7220). Cells were collected at 0 h, 12 h, 24 h, 48 h post serum deprivaton and lysed with RIPA lysis buffer. Each well of a 96 well plate contained 32 µl of caspase assay buffer, 2 µl of DMSO, 10 µl of 100 mM DTT, 20 µl of cell lysates (30 µg), 34 µl of distilled water and 2 µl of the 10 mM DEVD-aminoluciferin labeled with chromophore p-nitroaniline substrate (DEVD-pNA). After 4 h incubation at 37°C, the release of pNA from the substrate by caspase 3 (DEVDase) was detected using microtiter plate reader at 405 nm.
Apoptosis assay.
Cells showing apoptotic change were detected based on the translocation of membrane phosphatidyl-serine (PS) from the inner space to the surface of the cells. When PS is displayed externally, it can be detected by PS-affinitive Annexin V which has a fluorescent conjugate. The proportion of cells undergoing apoptosis in each cell line under both normal growing condition and following serum deprivation was quantified using Annexin V-Cy3 apoptosis detection kit (Biovision, #K102-100). After adding 5 µl of Annexin V-Cy3 into each cell suspension, apoptotic cells bound to Annexin V were detected by flow cytometry (BD FACSCalibur™ Flow Cytometer, BD Bioscience) at 570 nm emission wavelengths. In parallel, control cell suspension without Annexin V-Cy3 were also analyzed.
Statistical analysis.
All experiments were performed at least three times. Statistical analysis was performed by ANOVA and LSD (SPSS Software version 17.0, SPSS Inc.). Data were presented as mean ± standard deviation. Differences were considered to be significant if probability values of p < 0.05 or p < 0.01 were obtained.
Figures and Tables
Figure 1 Schematic features of miR cassettes and knockdown efficacy in N2a cells. (A) Each miR cassette in pcDNA6.2-GW/EmGFP vector. (B) Western blot of N2a cells transfected with each miR expression construct. (C) Quantitation of PrPC expression. When miRdual was introduced into the cells, PrPC expression was the most efficiently decreased as compared with wild-type cells (*p < 0.05, **P <0.01).
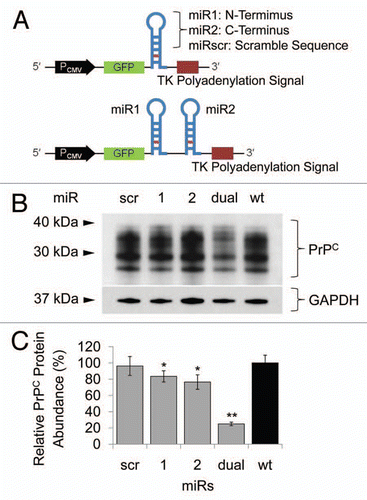
Figure 2 Transfection of miR cassettes and Prnp knockdown efficacy at mRNA and protein level. (A) PCR of genomic DNA shows miR cassette-specific amplicons. Lane L, 100 bp DNA ladder; lane 1, expression vector with miRdual cassette; lane 2, expression vector with miRscr; lane 3, wild-type cells; lane 4, N2amiRdual; lane 5, N2amiRscr; lane 6, no template. (B) qPCR analysis of PrPC transcript abundance showing knockdown efficiency of miRdual in mRNA levels. (C) Western blot analysis of PrPC protein abundance. Top part is the western blot and bottom panel is densitometric evaluation. dual1 and dual2 are N2amiRdual cell clones (*p < 0.05, **p < 0.01).
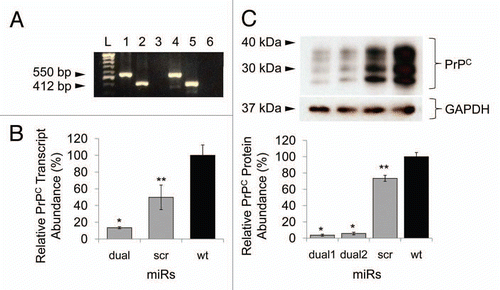
Figure 3 Quantitative PCR analysis of PrPC interacting and/or associated molecules. 6720460F02Rik, Plk3, Ppp2r2b, Csnk2a1 transcripts were upregulated in prion knockdown N2amiRdual cells compared to wild-type N2a cells. The increased Mpg transcripts in both N2amiRdual and N2amiRscr seemed to have no correlation with decreased PrPC (*p < 0.05, **p < 0.01).
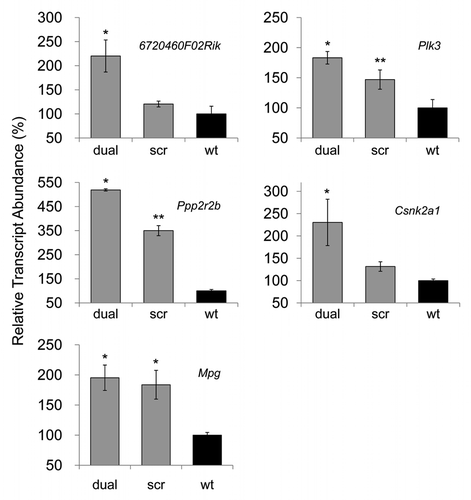
Figure 4 Cell proliferation, viability and GFP expression test. (A) Viability and proliferation of N2a cells stably transformed with miRdual or miRscr (*p < 0.05). (B) Confocal microscopy images of clonal expanded cells. N2amiRdual and N2amiRscr showed green fluorescent signals. Cell nuclei were stained with DAPI (blue). Photos were taken at 20 doubling passages. GFP signals stably expressed over 135 doubling passages. Scale bar, 50 um.
Figure 5 Apoptotic resistance to serum deprivation. Cells were cultured under conditions of serum deprivation and collected at indicated time points. (A) Detection of Drp1 expression levels analyzed by densitometric analysis of western blot. (B) Cytochrome C expression in cytosolic fractions detected by immunoassay. (C) Analysis of caspase 3 activity calculated by comparison with the free pNA. dual, N2amiRdual; scr, N2amiRscr; wt, miR non-treated wild-type N2a (*p < 0.05, **p < 0.01).
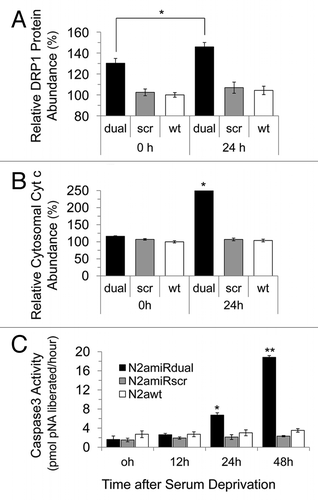
Figure 6 Detection of apoptotic cells following serum deprivation using Annexin V-Cy3 assay. Cells were collected at the indicated time points and 5 × 105 cell suspensions were treated with Annexin V. The proportion of cells showing apoptotic change was increased in N2amiRdual whereas most of cells showed the resistance against apoptosis stimulation in N2amiRscr and wild-type N2a cells (*p < 0.05).
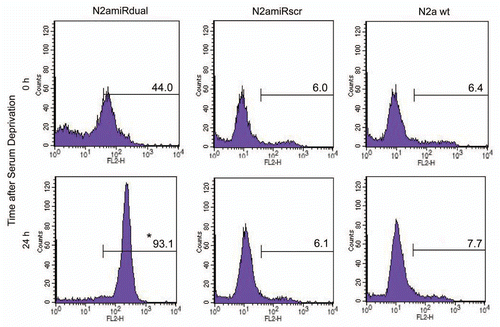
Figure 7 Downregulation pattern of PrPC in RK13-moPrnp cells harvested at different confluences. RK13-moPrnp cells were lentiviral-transduced with miR expression cassettes and the relative abundances of PrPC and GFP processed were determined by western blot (*p < 0.05, **p < 0.01).
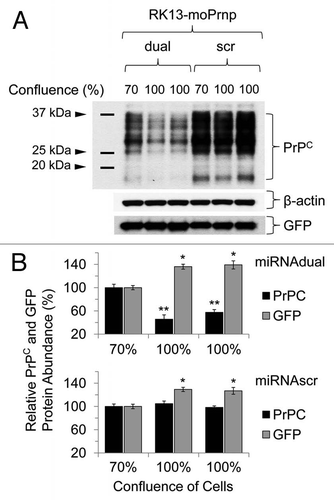
Table 1 Target genes of qPCR for characterizing the established N2amiRdual cell line
Table 2 Sequences of artificial miRs targeting Mus musculus Prnp ORF
Table 3 Primer sequences used in this study
Acknowledgments
This study was supported by BK21 for Veterinary Science, KRF-2006-005-J502901 and Research Institute of Veterinary Science, Seoul National University, Korea and by the Alberta Prion Research Institute for the work performed at the Centre for Prions and Protein Folding Diseases at the University of Alberta, Canada.
References
- Sutou S, Kunishi M, Kudo T, Wongsrikeao P, Miyagishi M, Otoi T. Knockdown of the bovine prion gene PRNP by RNA interference (RNAi) technology. BMC Biotechnol 2007; 7:44
- Prusiner SB. Prions. Proc Natl Acad Sci USA 1998; 95:13363 - 13383
- Basler K, Oesch B, Scott M, Westaway D, Walchli M, Groth DF, et al. Scrapie and cellular PrP isoforms are encoded by the same chromosomal gene. Cell 1986; 46:417 - 428
- Trevitt CR, Collinge J. A systematic review of prion therapeutics in experimental models. Brain 2006; 129:2241 - 2265
- Nuvolone M, Aguzzi A, Heikenwalder M. Cells and prions: a license to replicate. FEBS Lett 2009; 583:2674 - 2684
- Aguzzi A, Heikenwalder M. Prion diseases: Cannibals and garbage piles. Nature 2003; 423:127 - 129
- White MD, Mallucci GR. Therapy for prion diseases: Insights from the use of RNA interference. Prion 2009; 3:121 - 128
- Bueler H, Fischer M, Lang Y, Bluethmann H, Lipp HP, DeArmond SJ, et al. Normal development and behaviour of mice lacking the neuronal cell-surface PrP protein. Nature 1992; 356:577 - 582
- Nico PB, de-Paris F, Vinade ER, Amaral OB, Rockenbach I, Soares BL, et al. Altered behavioural response to acute stress in mice lacking cellular prion protein. Behav Brain Res 2005; 162:173 - 181
- Milhavet O, Casanova D, Chevallier N, McKay RD, Lehmann S. Neural stem cell model for prion propagation. Stem Cells 2006; 24:2284 - 2291
- Beranger F, Mange A, Solassol J, Lehmann S. Cell culture models of transmissible spongiform encephalopathies. Biochem Biophys Res Commun 2001; 289:311 - 316
- Mittal V. Improving the efficiency of RNA interference in mammals. Nat Rev Genet 2004; 5:355 - 365
- Aguzzi A, Sigurdson C, Heikenwaelder M. Molecular mechanisms of prion pathogenesis. Annu Rev Pathol 2008; 3:11 - 40
- Zomosa-Signoret V, Arnaud JD, Fontes P, Alvarez-Martinez MT, Liautard JP. Physiological role of the cellular prion protein. Vet Res 2008; 39:9
- Aguzzi A, Heikenwalder M. Pathogenesis of prion diseases: current status and future outlook. Nat Rev Microbiol 2006; 4:765 - 775
- Mallucci G, Collinge J. Rational targeting for prion therapeutics. Nat Rev Neurosci 2005; 6:23 - 34
- Spray DC, Iacobas DA. Organizational principles of the connexin-related brain transcriptome. J Membr Biol 2007; 218:39 - 47
- Insel PA, Patel HH. Do studies in caveolin-knockouts teach us about physiology and pharmacology or instead, the ways mice compensate for 'lost proteins'?. Br J Pharmacol 2007; 150:251 - 254
- Kim VN. Small RNAs: classification, biogenesis and function. Mol Cells 2005; 19:1 - 15
- Cullen BR. Enhancing and confirming the specificity of RNAi experiments. Nat Methods 2006; 3:677 - 681
- Whangbo JS, Hunter CP. Environmental RNA interference. Trends Genet 2008; 24:297 - 305
- Clark J, Ding S. Generation of RNAi libraries for high-throughput screens. J Biomed Biotechnol 2006; 2006:45716
- Vilette D, Andreoletti O, Archer F, Madelaine MF, Vilotte JL, Lehmann S, et al. Ex vivo propagation of infectious sheep scrapie agent in heterologous epithelial cells expressing ovine prion protein. Proc Natl Acad Sci USA 2001; 98:4055 - 4059
- Drisaldi B, Coomaraswamy J, Mastrangelo P, Strome B, Yang J, Watts JC, et al. Genetic mapping of activity determinants within cellular prion proteins: N-terminal modules in PrPC offset pro-apoptotic activity of the Doppel helix B/B' region. J Biol Chem 2004; 279:55443 - 55454
- Lee Y, Kim M, Han J, Yeom KH, Lee S, Baek SH, et al. MicroRNA genes are transcribed by RNA polymerase II. EMBO J 2004; 23:4051 - 4060
- Lewis BP, Shih IH, Jones-Rhoades MW, Bartel DP, Burge CB. Prediction of mammalian microRNA targets. Cell 2003; 115:787 - 798
- Detrait ER, Bowers WJ, Halterman MW, Giuliano RE, Bennice L, Federoff HJ, et al. Reporter gene transfer induces apoptosis in primary cortical neurons. Mol Ther 2002; 5:723 - 730
- Liu HS, Jan MS, Chou CK, Chen PH, Ke NJ. Is green fluorescent protein toxic to the living cells?. Biochem Biophys Res Commun 1999; 260:712 - 717
- Torbett BE. Reporter genes: too much of a good thing?. J Gene Med 2002; 4:478 - 479
- Satoh J, Obayashi S, Misawa T, Sumiyoshi K, Oosumi K, Tabunoki H. Protein microarray analysis identifies human cellular prion protein interactors. Neuropathol Appl Neurobiol 2009; 35:16 - 35
- Satoh J, Yamamura T. Gene expression profile following stable expression of the cellular prion protein. Cell Mol Neurobiol 2004; 24:793 - 814
- Meggio F, Negro A, Sarno S, Ruzzene M, Bertoli A, Sorgato MC, et al. Bovine prion protein as a modulator of protein kinase CK2. Biochem J 2000; 352:191 - 196
- Dong CF, Wang XF, Wang X, Shi S, Wang GR, Shan B, et al. Molecular interaction between prion protein and GFAP both in native and recombinant forms in vitro. Med Microbiol Immunol 2008; 197:361 - 368
- Archangelo LF, Greif PA, Holzel M, Harasim T, Kremmer E, Przemeck GK, et al. The CALM and CALM/AF10 interactor CATS is a marker for proliferation. Mol Oncol 2008; 2:356 - 367
- Zimmerman WC, Erikson RL. Polo-like kinase 3 is required for entry into S phase. Proc Natl Acad Sci USA 2007; 104:1847 - 1852
- Conn CW, Hennigan RF, Dai W, Sanchez Y, Stambrook PJ. Incomplete cytokinesis and induction of apoptosis by overexpression of the mammalian polo-like kinase, Plk3. Cancer Res 2000; 60:6826 - 6831
- Linden R, Martins VR, Prado MA, Cammarota M, Izquierdo I, Brentani RR. Physiology of the prion protein. Physiol Rev 2008; 88:673 - 728
- Knott AB, Perkins G, Schwarzenbacher R, Bossy-Wetzel E. Mitochondrial fragmentation in neurodegeneration. Nat Rev Neurosci 2008; 9:505 - 518
- Dagda RK, Merrill RA, Cribbs JT, Chen Y, Hell JW, Usachev YM, et al. The spinocerebellar ataxia 12 gene product and protein phosphatase 2A regulatory subunit Bbeta2 antagonizes neuronal survival by promoting mitochondrial fission. J Biol Chem 2008; 283:36241 - 36248
- Litchfield DW. Protein kinase CK2: structure, regulation and role in cellular decisions of life and death. Biochem J 2003; 369:1 - 15
- Chen J, Gao C, Shi Q, Wang G, Lei Y, Shan B, et al. Casein kinase II interacts with prion protein in vitro and forms complex with native prion protein in vivo. Acta Biochim Biophys Sin (Shanghai) 2008; 40:1039 - 1047
- Heriche JK, Lebrin F, Rabilloud T, Leroy D, Chambaz EM, Goldberg Y. Regulation of protein phosphatase 2A by direct interaction with casein kinase 2alpha. Science 1997; 276:952 - 955
- Kisby GE, Lesselroth H, Olivas A, Samson L, Gold B, Tanaka K, et al. Role of nucleotide- and base-excision repair in genotoxin-induced neuronal cell death. DNA Repair (Amst) 2004; 3:617 - 627
- Kisby GE, Olivas A, Park T, Churchwell M, Doerge D, Samson LD, et al. DNA repair modulates the vulnerability of the developing brain to alkylating agents. DNA Repair (Amst) 2009; 8:400 - 412
- Steele AD, Emsley JG, Ozdinler PH, Lindquist S, Macklis JD. Prion protein (PrPC) positively regulates neural precursor proliferation during developmental and adult mammalian neurogenesis. Proc Natl Acad Sci USA 2006; 103:3416 - 3421
- Wu G, Nakajima K, Takeyama N, Yukawa M, Taniuchi Y, Sakudo A, et al. Species-specific anti-apoptotic activity of cellular prion protein in a mouse PrP-deficient neuronal cell line transfected with mouse, hamster and bovine Prnp. Neurosci Lett 2008; 446:11 - 15
- Anantharam V, Kanthasamy A, Choi CJ, Martin DP, Latchoumycandane C, Richt JA, et al. Opposing roles of prion protein in oxidative stress- and ER stress-induced apoptotic signaling. Free Radic Biol Med 2008; 45:1530 - 1541
- Kim BH, Lee HG, Choi JK, Kim JI, Choi EK, Carp RI, et al. The cellular prion protein (PrPC) prevents apoptotic neuronal cell death and mitochondrial dysfunction induced by serum deprivation. Brain Res Mol Brain Res 2004; 124:40 - 50
- Frank S, Gaume B, Bergmann-Leitner ES, Leitner WW, Robert EG, Catez F, et al. The role of dynamin-related protein 1, a mediator of mitochondrial fission, in apoptosis. Dev Cell 2001; 1:515 - 525
- Breckenridge DG, Stojanovic M, Marcellus RC, Shore GC. Caspase cleavage product of BAP31 induces mitochondrial fission through endoplasmic reticulum calcium signals, enhancing cytochrome c release to the cytosol. J Cell Biol 2003; 160:1115 - 1127
- Abu-Qare AW, Abou-Donia MB. Biomarkers of apoptosis: release of cytochrome c, activation of caspase-3, induction of 8-hydroxy-2′-deoxyguanosine, increased 3-nitrotyrosine and alteration of p53 gene. J Toxicol Environ Health B Crit Rev 2001; 4:313 - 332
- Shigemura N, Kiyoshima T, Sakai T, Matsuo K, Momoi T, Yamaza H, et al. Localization of activated caspase-3-positive and apoptotic cells in the developing tooth germ of the mouse lower first molar. Histochem J 2001; 33:253 - 258
- Gottlieb RA, Granville DJ. Analyzing mitochondrial changes during apoptosis. Methods 2002; 26:341 - 347
- Li K, Li Y, Shelton JM, Richardson JA, Spencer E, Chen ZJ, et al. Cytochrome c deficiency causes embryonic lethality and attenuates stress-induced apoptosis. Cell 2000; 101:389 - 399
- McBride JL, Boudreau RL, Harper SQ, Staber PD, Monteys AM, Martins I, et al. Artificial miRNAs mitigate shRNA-mediated toxicity in the brain: implications for the therapeutic development of RNAi. Proc Natl Acad Sci USA 2008; 105:5868 - 5873
- Kang SG, Roh YM, Kang ML, Kim YS, Yoo HS. Mouse neuronal cells expressing exogenous bovine PRNP and simultaneous downregulation of endogenous mouse PRNP using siRNAs. Prion 4:32 - 37
- Smith C. Sharpening the tools of RNA interference. Natuer Methods 2006; 3:475 - 486
- Choi JK, Park SJ, Jun YC, Oh JM, Jeong BH, Lee HP, et al. Generation of monoclonal antibody recognized by the GXXXG motif (glycine zipper) of prion protein. Hybridoma 2006; 25:271 - 277