Abstract
The prion diseases occur following the conversion of the cellular prion protein (PrPC) into a disease-related isoform (PrPSc). In this study a cell painting technique was used to examine the role of the glycosylphosphatidylinositol (GPI) anchor attached to PrPC in prion formation. The introduction of PrPC to infected neuronal cells increased the cholesterol content of cell membranes, increased activation of cytoplasmic phospholipase A2 (cPLA2) and increased PrPSc formation. In contrast, PrPC with a monoacylated GPI anchor did not alter the amount of cholesterol in cell membranes, was not found within lipid rafts and did not activate cPLA2. Although monoacylated PrPC remains within cells for longer than native PrPC it was not converted to PrPSc. Moreover, the presence of monoacylated PrPC displaced cPLA2 from PrPSc-containing lipid rafts, reducing the activation of cPLA2 and PrPSc formation. We conclude that acylation of the GPI anchor attached to PrPC modifies the local membrane microenvironments that control some cell signaling pathways, the trafficking of PrPC and PrPSc formation. In addition, such observations raise the possibility that the pharmacological modification of GPI anchors might constitute a novel therapeutic approach to prion diseases.
A key event in the prion diseases is the conversion of a normal host protein (PrPC) into a disease-associated isoform (PrPSc).Citation1 Although the presence of PrPC is essential for prion formation,Citation2,Citation3 not all cells that express PrPC are permissive for PrPSc replication. The reasons why some cells that express PrPC do not replicate PrPSc are not fully understood. Reports that the targeting of PrPC to specific membranes is required for efficient PrPSc formationCitation4 indicate that the factors that affect the cellular targeting and intracellular trafficking of PrPC are critical in determining PrPSc replication.
Our study examined the effects of the glycosylphosphatidylinositol (GPI) anchor that links the majority of PrPC molecules to cell membranesCitation5 on PrPSc formation. Originally GPI anchors were seen as a simple method of attaching proteins to cell membranes. However, there is increasing interest in the role of GPI anchors in complex biological functions including the regulation of membrane composition, cell signaling and protein trafficking.Citation6 To examine the role of the GPI anchor PrPC preparations were digested with phosphatidylinositol-phospholipase C (PI-PLC) (deacylated PrPC) or phospholipase A2 (PLA2) (monoacylated PrPC) () and isolated by reverse phase chromatography. These digestions, coupled with a cell painting technique, allowed us to examine modifications of the GPI anchor that could not be achieved by genetic manipulation methods. Controversy surrounds the role of the GPI anchor in PrPSc formation; the seminal observation that transgenic mice producing anchorless PrPC produced large amounts of extracellular PrPSc,Citation7 suggests that the GPI has little effect upon PrPSc replication. In contrast, a recent study showed that cells that produce anchorless PrPC were not permissive to PrPSc formationCitation8 and in our study deacylated PrPC did not affect PrPSc production. Although at first glance these results appear contradictory, they may be explained by reference to the site of conversion of PrPC to PrPSc. Clearly anchorless PrPC can be converted to PrPSc in a process that occurs within the extracellular milieu. However, as anchorless PrPC is rapidly secreted from cellsCitation7 it has little contact with cell-associated PrPSc. Similarly we found that deacylated PrPC was fully soluble and did not readily associate with cells.
Native PrPC is rapidly transferred to cellsCitation9 and we showed that the addition of PrPC caused a dose-dependent increase in the PrPSc content of all prion-infected cell lines tested. We used this cell painting technique to introduce monoacylated PrPC to recipient cells. Our paper describes three major observations; firstly that monoacylated PrPC behaves differently to native PrPC with regards to cellular distribution, intracellular trafficking and cell signaling; secondly, that monoacylated PrPC was not converted to PrPSc and thirdly, that monoacylated PrPC inhibited the conversion of endogenous PrPC to PrPSc.
The presence of GPI anchors targets proteins including PrPC and PrPSc to specialized membrane micro-domains that are commonly called lipid rafts.Citation10,Citation11 Lipid rafts are patches of membranes that are highly enriched in cholesterol and sphingolipids and which are operationally defined by their insolubility in cold non-ionic detergents and floatation as low density membranes on sucrose density gradients. The importance of lipid rafts in prion diseases is based upon studies showing that treatment with cholesterol synthesis inhibitors reduced cellular cholesterol and the formation of PrPSc.Citation12 Since the cholesterol content of cell membranes is critical for the formation of lipid raftsCitation13 it is assumed that the integrity of these lipid rafts is necessary for efficient PrPSc formation. The presence of GPI-anchored proteins is thought to help lipid rafts form as the saturated fatty acids that are contained within GPI anchors facilitate the solubilisation of cholesterol in the membraneCitation14 and the glycan component protect cholesterol from water.Citation15 Thus the nature and number of the acyl chains contained within GPI anchors are factors that affect lipid raft formation ().
We observed that the addition of native PrPC significantly increased the amount of cholesterol in cell membranes. This result was unexpected as the amount of cholesterol in cell membranes is tightly controlled by an esterification and hydrolysis cycle which releases cholesterol from stored cholesterol esters.Citation16 The PrPC-induced increase in cholesterol was accompanied by a reduction in cholesterol esters suggesting that it was derived from the hydrolysis of cholesterol esters. Pharmacological inhibition of cholesterol ester hydrolysis not only blocked the PrPC-induced increase in cholesterol and the reduction in cholesterol esters, but also reduced the PrPC-induced increase in PrPSc formation. Collectively, these results indicate that cells respond to the introduction of PrPC by the hydrolysis of cholesterol esters; which provides cholesterol to stabilize PrPC within the lipid rafts that are necessary to facilitate PrPSc formation.
The differences in the cellular distribution of PrPC and monoacylated PrPC were examined using neurons from Prnp knockout mice. While PrPC was targeted to lipid rafts, monoacylated PrPC was found predominantly within the normal (non-raft) cell membranes. Unlike native PrPC, monoacylated PrPC did not affect the cholesterol content of cell membranes; an observation that highlights the critical role of the presence of two saturated fatty acids contained within the GPI anchor to sequester cholesterol and precipitate the formation of lipid rafts.Citation14 Critically, monoacylated PrPC was unable to solubilize cholesterol or precipitate lipid raft formation and was consequently found outside lipid rafts ().
Since many raft-associated proteins including PrPC traffic within cells via specific pathways,Citation17 we argued that monoacylated PrPC might undergo alternative trafficking pathways to those utilized by native PrPC. Our observations, that greater amounts of monoacylated PrPC than native PrPC were expressed at the cell surface, and that while most of the native PrPC was removed from these cells within 24 h, monoacylated PrPC remained in neurons for longer, are indicative of altered intracellular trafficking. It is possible that the cellular location and/or pathway(s) used by monoacylated PrPC may be physically segregated from PrPSc. This hypothesis would explain our observation that the addition of monoacylated PrPC to prion-infected cell lines did not increase PrPSc formation indicating that it was not converted to PrPSc.
Since monoacylated phospholipids exist only transiently within cell membranes, they are rapidly reacylated by esterases,Citation18 we wondered whether the monoacylated PrPC could also be reacylated to form the native, diacylated PrPC. We found no evidence that the monoacylated PrPC added to Prnp knockout neurons was reacylated suggesting that the enzymes involved in reacylation of membrane phospholipids do not recognize phosphatidylinositol when it is incorporated as part of the GPI anchor. In addition we were unable to detect monoacylated PrPC occurring naturally within Prnp wild-type neurons.
While these theories explain why monoacylated PrPC was not readily converted to PrPSc; a more refined hypothesis is required to explain why monoacylated PrPC reduced PrPSc production. One possibility is that monoacylated PrPC is converted to monoacylated PrPSc which in turn acts as an inefficient template for PrPC to PrPSc conversion.Citation19 It is also possible that monoacylated PrPC competes with endogenous PrPC for specific partner proteins involved in endocytosis. The depletion of these partner proteins could consequently alter the trafficking of endogenous PrPC and hence PrPSc formation. In our paper we explored the idea that the binding of monoacylated PrPC to PrPSc modifies the lipid rafts that are involved in PrPSc formation. Both the composition and function of lipid rafts is dynamic and controlled by an induced fit model.Citation20 Since monoacylated PrPC does not sequester cholesterol, the membrane surrounding a complex between PrPSc and monoacylated PrPC might be expected to contain less cholesterol than membranes formed following the interaction between PrPSc and PrPC (). Thus, the binding of monoacylated PrPC to PrPSc may reduce the cholesterol content of local membranes to a level below that required for the conversion of PrPC to PrPSc. This hypothesis is consistent with observations that formation of PrPSc was affected by the lipid composition of membranesCitation21 and that lipids were essential co-factors in prion formation.Citation22
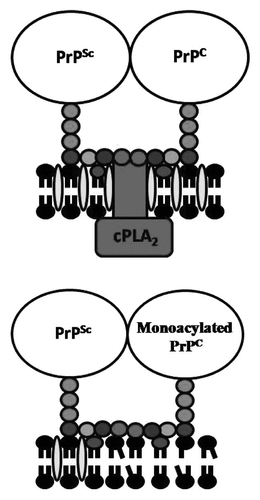
Our studies raise the question “why are lipid rafts important in PrPSc formation?” Lipid rafts are enriched with signaling molecules and can act as domains in which the GPI anchors attached to PrPC can interact with cell signaling pathways.Citation23 Although PrPC has been reported to interact with many cell signaling pathways we concentrated upon its effects on the activation of cPLA2, based upon studies showing that the activation of cPLA2 correlates strongly with the amounts of cholesterol and PrPSc,Citation24 and that the inhibition of cPLA2 reduces PrPSc formation.Citation25 These observations underpin the hypothesis that it is the clustering of GPI anchors attached to PrP proteins that leads to the activation of cPLA2. This hypothesis was tested by incubating Prnp knockout neurons with PrPC or monoacylated PrPC and then adding the anti-PrP mAb 4F2. We found that the cross-linkage of PrPC by mAb 4F2 caused the activation of cPLA2, whereas the cross-linkage of monoacylated PrPC by mAb 4F2 had no significant affect. The activation of cPLA2 is associated with multiple phosphorylation events and the translocation of cPLA2 to specific membranes.Citation26 In scrapie-infected GT1 (ScGT1) cells most of the cPLA2 was found within lipid rafts consistent with reports that the activation of cPLA2 is dependent upon cholesterol-sensitive lipid rafts.Citation27 More specifically, immunoprecipitation studies showed that cPLA2 was targeted to PrPSc-containing lipid rafts.Citation24 Collectively, these observations suggest that PrPC binds to PrPSc in cholesterol-dense lipid rafts, where it activates the cPLA2 that facilitates the conversion of PrPC to PrPSc ().
We found that the presence of monoacylated PrPC reduced the activation of cPLA2 within prion-infected cells. As cPLA2 can be activated by multiple different stimuli we sought to determine whether the inhibitory effect of monoacylated PrPC was stimulus specific. We reported that monoacylated PrPC did not affect the activation of cPLA2 by a phospholipase A2-activating peptide indicating that monoacylated PrPC did not have a direct inhibitory effect upon cPLA2. Rather we found that the addition of monoacylated PrPC to prion-infected cells caused the dissociation of some cPLA2 from PrPSc-containing lipid rafts. The targeting of cPLA2 to membranes containing their endogenous substrates can regulate cell signaling, including for the formation of second messengers such as platelet-activating factor that facilitate PrPSc formation.Citation25 We propose that the binding of monoacylated PrPC to PrPSc changed the composition of the underlying membrane so that it no longer captured and activated cPLA2 ().
In conclusion our study showed that the addition of monoacylated PrPC modified cell membranes thus reducing the activation of cPLA2 and PrPSc formation in prion-infected cells. We propose that the 2 acyl chains attached to the GPI anchor is a critical factor that facilitates the conversion to PrPC to PrPSc within cell membranes and that the presence of monoacylated PrPC disrupts lipid raft micro-domains that are essential for efficient PrPSc formation. Moreover, these results raise the possibility that targeting the GPI anchor attached to PrPC may reveal novel therapeutics/treatments for prion diseases.
Figures and Tables
Figure 1 Phospholipase digestion of PrPC affects the acylation of the GPI anchor. Cartoon showing the putative GPI anchor attached to PrPC, monoacylated PrPC and deacylated PrPC. Glycan residues shown include inositol (Inos), mannose (Man), sialic acid (SA), galactose (Gal), N-acetyl galactosamine (GalNAc) and glucosamine (GlcN) as well as phosphate (P).
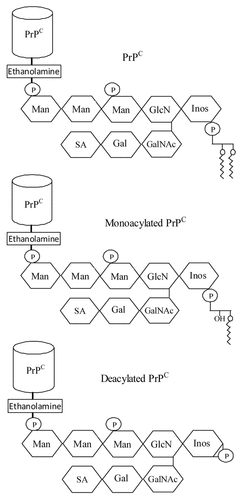
Figure 2 Acylation of PrPC affects the underlying cell membrane. Cartoon showing the proposed membranes surrounding native PrPC and monoacylated PrPC, including cholesterol (), lyso-phospholipids (![]()
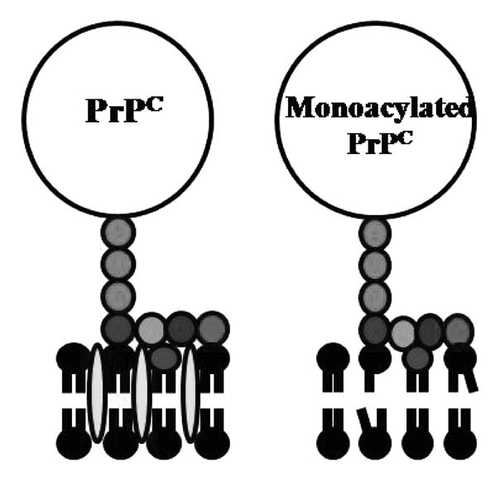
Figure 3 Monoacylated PrPC affects the capture of cPLA2 in PrPSc-containing lipid rafts. (A) Cartoon showing the proposed membranes surrounding PrPSc and PrPC including the capture of cPLA2 in lipid rafts that are dense in cholesterol (![]()
Acknowledgments
This work was supported by a grant from the European Commission FP6 “Neuroprion” Network of Excellence.
References
- Prusiner SB. Prions. Proc Natl Acad Sci USA 1998; 95:13363 - 13383
- Bueler H, Aguzzi A, Sailer A, Greiner RA, Autenried P, Aguet M, et al. Mice devoid of PrP are resistant to scrapie. Cell 1993; 73:1339 - 1347
- Mallucci G, Dickinson A, Linehan J, Klohn PC, Brandner S, Collinge J. Depleting neuronal PrP in prion infection prevents disease and reverses spongiosis. Science 2003; 302:871 - 874
- Beranger F, Mange A, Goud B, Lehmann S. Stimulation of PrP(C) retrograde transport toward the endoplasmic reticulum increases accumulation of PrP(Sc) in prion-infected cells. J Biol Chem 2002; 277:38972 - 38977
- Bate C, Williams A. Monoacylated cellular prion protein modifies cell membranes, inhibits cell signaling and reduces prion formation. J Biol Chem 2011; 286:8752 - 8758
- Chatterjee S, Mayor S. The GPI-anchor and protein sorting. Cell Mol Life Sci 2001; 58:1969 - 1987
- Chesebro B, Trifilo M, Race R, Meade-White K, Teng C, LaCasse R, et al. Anchorless prion protein results in infectious amyloid disease without clinical scrapie. Science 2005; 308:1435 - 1439
- McNally KL, Ward AE, Priola SA. Cells expressing anchorless prion protein are resistant to scrapie infection. J Virol 2009; 83:4469 - 4475
- Liu T, Li R, Pan T, Liu D, Petersen RB, Wong BS, et al. Intercellular transfer of the cellular prion protein. J Biol Chem 2002; 277:47671 - 47678
- Sotgia F, Razani B, Bonuccelli G, Schubert W, Battista M, Lee H, et al. Intracellular retention of glycosylphosphatidyl inositol-linked proteins in caveolin-deficient cells. Mol Cell Biol 2002; 22:3905 - 3926
- Vey M, Pilkuhn S, Wille H, Nixon R, DeArmond SJ, Smart EJ, et al. Subcellular colocalization of the cellular and scrapie prion proteins in caveolae-like membranous domains. Proc Natl Acad Sci USA 1996; 93:14945 - 14949
- Taraboulos A, Scott M, Semenov A, Avrahami D, Laszlo L, Prusiner SB, et al. Cholesterol depletion and modification of COOH-terminal targeting sequence of the prion protein inhibit formation of the scrapie isoform. J Cell Biol 1995; 129:121 - 132
- Brown DA, London E. Structure and function of sphingolipid- and cholesterol-rich membrane rafts. J Biol Chem 2000; 275:17221 - 17224
- Schroeder R, London E, Brown D. Interactions between saturated acyl chains confer detergent resistance on lipids and glycosylphosphatidylinositol (GPI)-anchored proteins: GPI-anchored proteins in liposomes and cells show similar behavior. Proc Natl Acad Sci USA 1994; 91:12130 - 12134
- Huang J, Feigenson GW. A microscopic interaction model of maximum solubility of cholesterol in lipid bilayers. Biophys J 1999; 76:2142 - 2157
- Chang TY, Chang CCY, Cheng D. Acyl-coenzyme A:cholesterol acyltransferase. Ann Rev Biochem 1997; 66:613 - 638
- Nichols B. Endocytosis of lipid-anchored proteins: excluding GEECs from the crowd. J Cell Biol 2009; 186:457 - 459
- Farooqui AA, Horrocks LA, Farooqui T. Deacylation and reacylation of neural membrane glycerophospholipids. J Mol Neurosci 2000; 14:123 - 135
- Bate C, Tayebi M, Williams A. The glycosylphosphatidylinositol anchor is a major determinant of prion binding and replication. Biochem J 2010; 428:95 - 181
- Pike LJ. Lipid rafts: heterogeneity on the high seas. Biochem J 2004; 378:281 - 292
- Graham JF, Agarwal S, Kurian D, Kirby L, Pinheiro TJT, Gill AC. Low density subcellular fractions enhance disease-specific prion protein misfolding. J Biol Chem 2010; 285:9868 - 9880
- Wang F, Wang X, Yuan CG, Ma J. Generating a prion with bacterially expressed recombinant prion protein. Science 2010; 327:1132 - 1135
- Simons K, Toomre D. Lipid rafts and signal transduction. Nat Rev Mol Cell Biol 2000; 1:31 - 39
- Bate C, Tayebi M, Williams A. Sequestration of free cholesterol in cell membranes by prions correlates with cytoplasmic phospholipase A2 activation. BMC Biol 2008; 6:8
- Bate C, Reid S, Williams A. Phospholipase A2 inhibitors or platelet-activating factor antagonists prevent prion replication. J Biol Chem 2004; 279:36405 - 36411
- Nalefski EA, Sultzman LA, Martin DM, Kriz RW, Towler PS, Knopf JL, et al. Delineation of two functionally distinct domains of cytosolic phospholipase A2, a regulatory Ca(2+)-dependent lipid-binding domain and a Ca(2+)-independent catalytic domain. J Biol Chem 1994; 269:18239 - 18249
- Gaudreault SB, Chabot C, Gratton JP, Poirier J. The caveolin scaffolding domain modifies 2-amino-3-hydroxy-5-methyl-4-isoxazole propionate receptor binding properties by inhibiting phospholipase A2 activity. J Biol Chem 2004; 279:356 - 362