Abstract
The “protein only” hypothesis states that the key phenomenon in prion pathogenesis is the conversion of the host protein (PrPC) into a b-sheet enriched polymeric and pathogenic conformer (PrPSc). However the region of PrP bearing the information for structural transfer is still controversial. In a recent report, we highlighted the role of the C terminal part i.e. the helixes H2 and H3, using mutation approaches on recombinant PrP. The H2H3 was shown to be the minimal region necessary to reproduce the oligomerisation pattern of the full-length protein. The oligomers produced from isolated H2H3 domain presented the same structural characteristics as the oligomers formed from the full-length PrP. Combining other groups’ results, this paper further discusses the relative, direct or indirect role of different PrP regions in assembly. The H2H3 region represents the core of PrP oligomers and fibrils, whereas the N terminus could explain divergences among different aggregates. Finally this review evocates the possibility to separate the domain involved in prion information transference (i.e. prion replication) from the domain bearing the cytotoxicity properties.
Transmissible spongiform encephalopathies (TSE), fatal neurodegenerative diseases affecting humans and other mammalians, induce in most cases loss of motor control and dementia. PrP is a protein physiologically present in parts of the animal kingdom (in mammals, birds, reptiles and fishes). According to the “protein-only” hypothesis,Citation1,Citation2 the key phenomenon in the pathogenesis is the conversion of the α-helix rich host-encoded PrP form (PrPC) into a pathogenic conformer (PrPSc) characterized by a higher content in β-sheet and a polymeric state. The conversion to an enriched β-sheet structure is supposed to be due to the modification—induced only by a PrPSc-like state acting as a template- of PrPC into the PrPSc conformer. This hypothesis was first proposed by Griffith in 1967Citation3 and revisited by Lansbury et al. in 1993.Citation4 The prion hypothesis has now found increasing support from experimental evidence based on the synthetic production of β-sheeted recombinant PrP which shows pathogenic properties in a wide variety of physico-chemical conditions.Citation5–Citation7 However, the molecular basis of prion conversion remains unclear, especially the various structural landscape of the PrPSc, which is the basis of the strain phenomenon.Citation8
To understand the mechanisms of transfer of the structural information, two mains issues have to be addressed: (1) we need to understand which region(s) of the protein act as template for conversion and (2) what is the “pathogenic” state of this domain. In this review, we shall assume that the region bearing the infectious information for replication and the region responsible for polymerization are identical. However, the link between the propensity of a domain to form aggregates and the ability to contain the necessary information for prion replication is far from being trivial. Generally the formation of amyloid assemblies results from the aggregation of disordered peptides or in some cases from disordered regions of a folded protein.Citation8 If we consider that prion replication is only supported by the globular part of PrPCitation9 the currently available model involves the folded domain. Since all structural transitions need at least a partial unfolding and refolding process, pre-required structural events should be considered prior to the conversion process.
PrP Structural Dynamic and S2H2 Loop
The subcellular localization sites of prion replication are not yet well established. It was reported that the conversion process could take place either at the cell surface at the vicinity of the cell membrane below the neutral pH, or in the endosomes at pH 4.7–5.8. At pH 4.5, the region 154–157 (C-terminal of helix H1) and region 161–164 (β-strand S2 region) have a larger tendency to unfold than at pH 7.0 (human numbering).Citation10 Indeed, the aggregation pathway of PrP depends on pH. reports the quaternary structure of OvPrP (VRQ variant) at different pH values after incubation at 50°C. At pH below 4.5 and above 6.0 the main stable fold of PrP corresponds to a β enriched oligomeric state whereas at pH 4.5 oligomerization is almost suppressed. Oligomer dispersion is also affected by pH variation. At neutral pH mainly the largest type of oligomers (O1) is produced whereas at pH 3.5 discreet and smaller oligomers (O3) are formed in a parallel pathway to the O1 oligomers.Citation11 According to crystallographic and NMR structures, the charged residues (Asp, Glu and His) are mostly located on the PrP surface. This localization leads to admit quasi-equivalence between theoretical and experimental pK and proposes a correlation between the environmental pH and the protonation state of Asp, Glu and His residues. Indeed, in the pH range (pH > 6.5) in which His residues are de-protonated partial unfolding of PrP leads to the irreversible formation of O1 oligomers. The same process takes place when Glu residues are in a protonated state (pH < 4.2). The conformational landscape of PrP increases with the apparition of a new oligomer (called O3) when the Asp residues start to be protonated. These results underline that conformational diversity of PrP and folding are affected by environmental factors.
The loop between S2 and helix H2 is one of the regions of PrP which is in the spotlight for the conversion process. Kaneko and colleagues report this region as a binding site for factor X, a protein of yet unknown nature suggested to be a PrP binding partner.Citation12 In sheep PrP a polymorphism at position 171 (sheep numbering) controls sensitivity to scrapie agent. Several NMR studies reported the S2H2 region as a “switch” region controlling the species barrierCitation13 with a crucial role of positions 170 (S or N) and 174 (N or T).Citation14
In order to determine the structural dynamics of the S2H2 loop in PrP conformational changes, different covalent constraints were introduced by engineering a disulfide bridge in full-length sheep PrP. These covalent constraints have the effect of reducing specific breathing modes of the native state.Citation11 The double mutants Y160C M209C (sheep numbering) covalently attaches the H1S2 loop to helix H3 and prevents the unfolding of H2H3 domain from the S1H1S2 region. When this mutant was tested for aggregation either by thermal perturbationCitation15 or by chaotropic treatments,Citation6,Citation16 no conversion was observed ( and B). Moreover, the dynamic of the S2H2 loop was also shown to play an important role in the polymerization pathway and oligomer conformational diversity. Indeed, when the mobility of S2H2 is reduced through a disulfide bond between the S2H2 loop and the H3 region (V169C E224C) (sheep numbering) the formation of O3 oligomers is drastically reduced (). A more recent paper indicates that only disulfides which tether S1H1S2 to H2H3 prevented PrP conversion in vitro and in prion infected cell cultures.Citation17 These observations suggest that the event required prior polymerization consists in the separation of H2H3 domain from the rest of the protein (). Therefore the structural perturbation of the S2H2 loop constitutes a key step for further structural evolution and conformational transition and any mutation or factor binding to this domain should then affect the rate and pathway of the conversion.
Domain Involved in Prion Structural Information Transference
The prion hypothesis proposed by PrusinerCitation2 is based on structural information transfer (SIT) which involves a common mechanism for protein/protein interaction described by Koshland and called “induced fit” hypothesis.Citation18 The molecular basis of SIT explicitly needs a region of PrP serving as a template. Some models consider the S1H1S2 region as the region serving as template, whereas other models involve the H2H3 region. Wille and co-workers have constructed a model of PrPSc based on electron diffraction that suggests S1H1S2 as the region involved in PrPSc formation.Citation19 According to the Wille's model, the H2H3 domain of PrP remains in an α-helical structure and the disulfide bridge is not disrupted. Wille and co-workers' model is consistent with Come and colleagues' model proposed in 1993.Citation4 According to this model the conversion of PrPC to PrPSc occurs through the elongation of S1 and S2. Later Morrissey and Shakhnovich underlined the unusual hydrophilic character of H1 and proposed that it could be involved in the structural transition.Citation20 These observations suggest a model in which S1 and S2 serve as the seed for β strand elongation leading to H1 unfolding (). This model is consistent also with the widespread idea that participation of the short S1S2 β-sheet is crucial for seeding β-sheet elongation as well as the presence of a “steric zipper” stretch.Citation21 Surewicz's group reported that the PrP fragment 23–144 which is generated by Y145Stop mutation in human PrP is able to form amyloid fibrils.Citation22 However, transmission from Y145Stop animals to other model animals was so far unsuccessful.Citation23
Several observations converge onto a role of H2H3 in the PrP aggregation mechanism. From bioinformatics and experimental procedures, the peptide SNQNNF located in the loop between S2 and helix H2 and the beginning of H2 was shown to be a potential steric zipper leading to pair-of-sheet motifs and presumably to fibrils.Citation21 Khan and collaboratorsCitation24 underlined that the interaction between residue 171 in the same region and the serine residue present in rabbit PrP at position 174 (human numbering) induces a helix capping that stabilises the N terminus of H2. The authors proposed that this could explain the low susceptibility of rabbit PrP for prion diseases compared to other mammals. Intra-chain distance estimation on tagged PrP amyloid fibrils obtained under chaotropic treatment is in favor of the involvement of the helices H2 and H3 in large amyloid formation.Citation25,Citation26 Furthermore replica exchange molecular dynamic simulations suggest that H2 and H3 form the stable core of a partially unfolded candidate for aggregation whereas H1 has a higher mobility.Citation27
It was shown previously that recombinant full-length ovine PrP protein (OvPrP) forms discrete soluble oligomeric species upon thermal unfolding.Citation11 These species are formed along a broad temperature range (20–85°C) with ratios depending on the temperature. They are induced by three independent kinetic pathways leading to three types of oligomers O1, O2 and O3 (as observed by size-exclusion chromatography) with different stability against heating (O1 being the most stable and O2 the less stable).
A protein corresponding to the region 167–234 (spanning the whole H2H3 domain) of ovine PrP (sheep numbering) refolds after purification into a α-helical rich peptide as judged by CD.Citation28 NMR investigations reveal a similar fold (RMSD <1.5 Å) for the isolate H2H3 domain and for H2H3 in the full length context.Citation29 Upon heating at 20°C or above, the ovine H2H3 protein forms discrete oligomers with a pattern similar to the one observed for full-length OvPrP. The same observation was confirmed with the mouse and human H2H3 domain. Analysis of the rate of depolymerization of purified oligomers obtained from the H2H3 domain and full-length PrP reveals that N-terminus and the S1H1S2 region do not influence oligomer stability.Citation28 Taken together, these observations indicate that H2H3 constitutes a minimal domain supporting oligomerization diversity.
The H2H3 domain is not only involved in oligomerization but also constitutes the core of fibrils. Lu and colleagues demonstrated by hydrogen/deuterium exchange that H2H3 may be involved in PrP amyloid fibrils obtained after chaotropic treatment.Citation30 Further investigations using site directed spin labelling and solid state NMR of PrP fibrils obtained under the same conditions led to propose a model in which H2H3 is involved in amyloid formation.Citation25,Citation31 More recently the bistability of H2H3 was shown in experiments in which H2H3 in an α-fold undergoes a structural transition without any treatment to form amyloid fibrils.Citation29 This bistability makes H2H3 unique as compared to different yeast prions for which the prion domain is a disordered region.Citation32
Selection of the Oligomerization and PrP Conformational Landscape
Several oligomerization pathways of PrP may coexist.Citation11,Citation33 To test which pathway(s) can generate fibrils, a critical concentration of the amyloidogenic precursor needs to be overcome, as for other amyloidogenic proteins. Upon concentration by ultracentrifugation, we previously have shown that the larger type of oligomers, O1, assembles into fibrils, in contrast to O3 which constitutes a dead end pathway.Citation11 The aggregation pathway plays an important role in prion disease as it is commonly accepted that the species barrier and strain phenomenon are both due to different conversion pathways.Citation34 In contrast to ovine PrP, mouse and hamster PrP form only O3 oligomers.Citation28 To test whether H2H3 harbors in its sequence the required information for the oligomerization pathway selection we have tried to reproduce the polymerization pattern of mouse PrP by single mutations introduced in the ovine PrP scaffold.Citation28 The polymorphism at position 208 of H3 (ovine numbering) constitutes the basis of the difference between ovine and mouse PrP oligomerization. Indeed the substitution of I208M in ovine H2H3 induces the mouse PrP pattern i.e., presence of O3 and absence of O1 oligomers. Moreover, alanine replacement of H190 (ovine numbering) in H2, the residue facing residue I208 in H3, inhibits formation of O3 oligomers and leads to selection of the O1 oligomerization pathway. From depolymerisation kinetics, the O3 oligomer from I208M and O3 from OvPrP present an isokinetic behaviour. A similar observation was made for O1 of H190A and OvPrP. These observations indicate that the difference in the oligomerization patterns does not come from a difference in the stability of the observed oligomers of the mutants but is related either to the enhancement of a oligomer pathway or to inhibition of the other oligomer pathway. Since I208M forms O3 at similar amounts as OvPrP (about 65% of the monomer after 60 min at 50°C), without modification of the O3 stability, this means that the mutation I208M directly inhibits O1 pathway. These results underline the fact that single mutations located on H2H3 lead to the selection of the polymerization pathway.
Monomer Destabilization Leads to PrP Activation
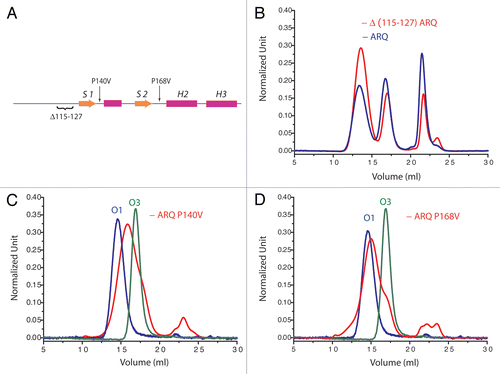
Considering in vivo conditions, a usual hypothesis states that conversion of PrP and some other amyloid-type proteins would occur by interactions with membranes. Indeed membranes offer both destabilization of proteins, by changes in physico-chemical local environment, and a decrease of the energy barrier for protein nucleation, by potential high local concentration of proteins which kinetically enhanced the rate of multi-order kinetics.Citation35 Independently of its GPI (glycophosphatidyl inositol) anchor, PrP binds to certain type of lipids. The binding of the N-terminal part of PrP to phosphatidylserine modifies PrP structure and destabilizes the C-terminal part.Citation36 This implicates a transmission of the unfolding signal through the N-terminal part to the rest of the protein. In presence of phosphatidylglycine (POPG) and RNA, Wang and co-workers were able to convert recombinant PrP into a highly pathogenic form.Citation37 The interaction with POPG is realized via the N-terminal positively charged domain and a “hydrophobic” region encompassing residues 100–134 (human numbering) including a cluster of lysines, a poly Ala stretch and the S1 region.Citation38 The interaction of the N-terminal hydrophobic domain was reported to be necessary for lipid inducing conversion and PK resistance of the PrP. However the oligomerization pattern of OvPrP (ARQ variant) is not affected by the deletion of region 115–127 (sheep numbering) encompassing the poly Ala stretch ( and B) even if the kinetics of aggregation is faster. Taking together these two results suggests that the N-terminal part may induce the transfer of an unfolding signal to the rest of the PrP, under certain conditions, without being part of the final product core.
The nature of the unfolding information that is transmitted to the rest of PrP leads to the exposure of the H2H3 domain and induces the activated form of PrP. Indeed it can be supposed that the N-terminal part and especially S1H1S2 constitute a steric hindrance for H2H3 oligomerization and need to be displaced (). Membrane interactions via the N-term part of PrP including S1 region might destabilize S1H1S2 and then the separation of this last region from H2H3. The following sequence could occur, according to molecular dynamics simulation (at 55°C) on a short H2H3 construct spanning residues C182 until C217 (sheep numbering). In this study the H2 unfolds while H3, more stable, unfolds only partly and progressively from M216 to T204 (sheep numbering).Citation28 The unfolding of H3 is accompanied by the refolding of the entire structure into a β hairpin. An alternative hypothesis for H2H3 activation (other than membrane interactions with N terminal part), is the involvement of a putative factor X that is proposed to interact with S2H2 loop.Citation12 It remains to be clarified which region(s) of H2H3 region of one PrP molecule further interacts with other H2H3 region(s).
We reported that the oligomerization pathway is affected by single mutations located on H2H3 domain. Although the same polymerization pattern was observed for purified H2H3 isolated domain and for H2H3 in the full-length protein, some of our unpublished data suggest that mutations in S1H1S2 region could also affect the oligomerization pathway. Indeed residue mutations as P140V or P168V (sheep numbering) lead to restrict the oligomerization pathway to the formation of O2 oligomers (, C and D). This observation suggests a potential role of S1H1S2 in either unfolding pathway or in the aggregation root even if the region 23–167 (sheep numbering) is not involved in the core of oligomers.
Most mutations leading to familial Creutzfeldt Jakob disease are located in the H2H3 region whereas most mutations leading to Gerstmann-Straüssler-Scheinker (GSS) disease are in the N terminal S1H1S2 regions.Citation39 A first explanation for the involvement of two regions could be that the domain involved in prion replication and the domain responsible for cytotoxicity would differ. Another possibility would be that the conformational dynamic of the rest of the protein outside of H2H3 would affect the polymerization pathway through different mechanisms (e.g., annealing or docking).
Conclusion
The aggregation of H2H3 seems to require the unfolding of S1H1S2. In this scheme the S2H2 loop plays a crucial role as it constitutes a hinge-loop between these two domains. For drug design, a strategy based on the stabilization of the native state (i.e., PrPC) should constitute a more suitable target than targeting PrPSc or PrP amyloid fibrils, which present an exceptional thermodynamic stability. Recently we have found that poly-D-lysine binds to PrP by overlapping H2H3 and H1. This binding leads to removal of PK-resistant PrP from SN56 chronically infected cells (Xu Z, et al. accepted in FASEB J.). Another molecule, GN8, which is able to bind H2, the H2-H3 loop and the H1-S2 loop, has been shown to prolong the survival of TSE-infected mice.Citation40
As underlined in the present report, domains of PrP other than H2H3 may also play important roles in the PrP conversion kinetics even if they are not directly involved as an aggregation domain. Another domain that we did not address in the present paper is the glycan domain of PrP. As glycosylation sites are located in H2H3, the packing of H2H3 in oligomers might explain a preferential selection of certain glycosylation types during PrP conversion. However the different ratios in PrPsc glycosylated forms (un-, mono- and diglycosylated) observed as a function of disease's subtype may be due mainly to differences in trafficking or in cell types.Citation41
An important issue that has to be considered is whether the domain of prion replication and the domain involved in the cytopathogenic events are identical. If we consider that prion cytotoxicity occurs through oligomers/membrane interaction,Citation42–Citation44 then the region directly involved in the cytopathogenicity process is the N terminal domain and therefore differs from the replication domain (H2H3). As the prion strain characterization is mostly based on the cytopathogenic criteria on inoculated animals (time of incubation, anathomopathologic profile etc.…), the concept of strain variability is then defined rather by the conformational diversity of the domain involved in cytotoxicity process than by the conformational diversity of the replication domain.
Abbreviations
| PrP | = | prion protein |
| PrPC | = | α-helix rich host-encoded PrP (“cellular” PrP) |
| PrPSc | = | β-sheet enriched polymeric conformer (PrP “scrapie”) |
| OvPrP | = | ovine PrP |
| GSS | = | Gerstmann-Straüssler-Scheinker disease |
| PK | = | proteinase K |
Figures and Tables
Figure 1 pH effect on the oligomerization pattern of V136 R154 Q171 (sheep numbering) PrP sheep variant from size-exclusion chromatography (after heating for 30 min 80 µM of protein at 50°C), following the methods already described in reference Citation11. At pH below 3.5 (blue line) the amount of O3 oligomers increases whereas at pH 4.1 (black line) and 7.15 (red line) the O1 oligomers are the most representative object. At pH 4.5 (green line) no oligomers were detected.
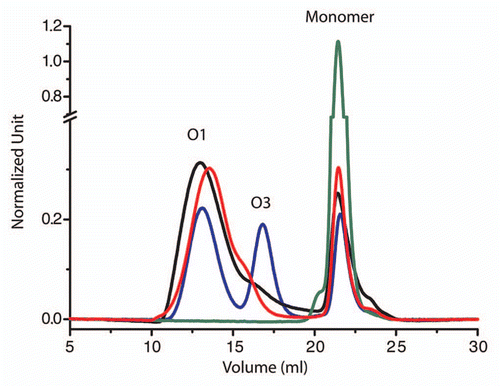
Figure 2 Effect of structural constraint on ovine PrP by additional disulfide bridge on oligomerization process (after heating for 90 min 80 µM of protein at 50°C). (A) Localization of the SS bounds on PrP structure. Left: Y160C M209C, right: V169C E224C (sheep numbering). (B) Their effect on the oligomerization process (black curves: wild-type ovine PrP (ARQ sheep variant); red curves: Y160C M209C and V169C E224C).
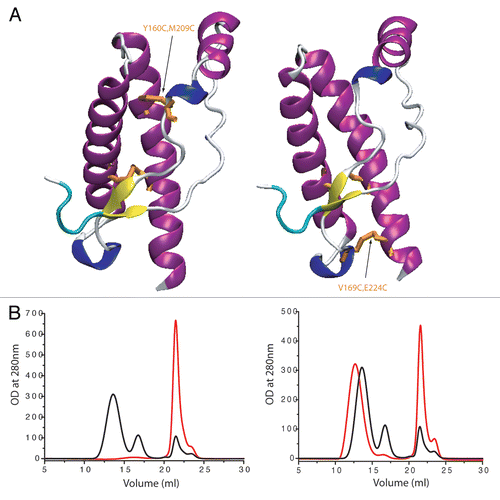
Figure 3 Prion structural dynamics in relation with polymerization process: mechanism of separation of S1H1S2 from H2H3. According to experiments with artificial disulfide bridge, H/D exchange and the fact that H2H3 summarizes the oligomerization pattern of full-length PrP, the activated state prior to aggregation corresponds to the separation of S1H1S2 from H2H3.
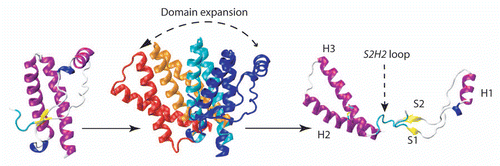
Figure 4 The structural basis of PrPC-to- PrPSc conversion according to S1S2 seeding model. The two β-strands S1 and S2 were proposed to play the role of “seed” for an elongation during the H1 unfolding.Citation44
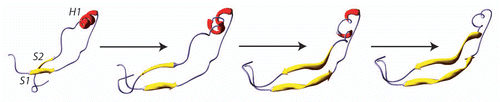
Figure 5 Deletion and mutations in N-terminal and S1H1S2 region affects oligomerization pattern. (A) The localization of the deletion and mutations on PrP structure. Size-exclusion chromatographs of (B) D(115–127) ARQ, (C) ARQ 140V and (D) ARQ P168V (sheep numbering). On chromatographs (C and D) is indicated the position of purified O1 (blue line) and the one of O3 oligomers (green line) from ARQ wild type. These observations indicate that mutations on S1H1S2 affect also the oligomerization pathway.
Acknowledgments
Financial support for this work was provided by the French National Institute of Agronomic Research (INRA) including INRA-Package and Fondation Alliance Biosecure. The authors thank Annalisa Pastore (MRC, London) and Muhammad K. Salamat (INRA, Jouy-en-Josas) for improving the manuscript.
References
- Prusiner SB. Novel proteinaceous infectious particles cause scrapie. Science 1982; 216:136 - 144
- Prusiner SB. Prions. Proc Natl Acad Sci USA 1998; 95:13363 - 13383
- Griffith JS. Self-replication and scrapie. Nature 1967; 215:1043 - 1044
- Come JH, Fraser PE, Lansbury PT Jr. A kinetic model for amyloid formation in the prion diseases: importance of seeding. Proc Natl Acad Sci USA 1993; 90:5959 - 5963
- Jackson GS, Hosszu LL, Power A, Hill AF, Kenney J, Saibil H, et al. Reversible conversion of monomeric human prion protein between native and fibrilogenic conformations. Science 1999; 283:1935 - 1937
- Swietnicki W, Morillas M, Chen SG, Gambetti P, Surewicz WK. Aggregation and fibrillization of the recombinant human prion protein huPrP90-231. Biochemistry 2000; 39:424 - 431
- Rezaei H, Eghiaian F, Perez J, Doublet B, Choiset Y, Haertle T, et al. Sequential generation of two structurally distinct ovine prion protein soluble oligomers displaying different biochemical reactivities. J Mol Biol 2005; 347:665 - 679
- Chiti F, Dobson CM. Amyloid formation by globular proteins under native conditions. Nat Chem Biol 2009; 5:15 - 22
- Shmerling D, Hegyi I, Fischer M, Blattler T, Brandner S, Gotz J, et al. Expression of amino-terminally truncated PrP in the mouse leading to ataxia and specific cerebellar lesions. Cell 1998; 93:203 - 214
- Calzolai L, Zahn R. Influence of pH on NMR structure and stability of the human prion protein globular domain. J Biol Chem 2003; 278:35592 - 35596
- Eghiaian F, Daubenfeld T, Quenet Y, van Audenhaege M, Bouin AP, van der Rest G, et al. Diversity in prion protein oligomerization pathways results from domain expansion as revealed by hydrogen/deuterium exchange and disulfide linkage. Proc Natl Acad Sci USA 2007; 104:7414 - 7419
- Kaneko K, Zulianello L, Scott M, Cooper CM, Wallace AC, James TL, et al. Evidence for protein X binding to a discontinuous epitope on the cellular prion protein during scrapie prion propagation. Proc Natl Acad Sci USA 1997; 94:10069 - 10074
- Perez DR, Damberger FF, Wuthrich K. Horse prion protein NMR structure and comparisons with related variants of the mouse prion protein. J Mol Biol 2010; 400:121 - 128
- Sigurdson CJ, Nilsson KP, Hornemann S, Manco G, Fernandez-Borges N, Schwarz P, et al. A molecular switch controls interspecies prion disease transmission in mice. J Clin Invest 2010; 120:2590 - 2599
- Rezaei H, Choiset Y, Eghiaian F, Treguer E, Mentre P, Debey P, et al. Amyloidogenic unfolding intermediates differentiate sheep prion protein variants. J Mol Biol 2002; 322:799 - 814
- Baskakov IV, Legname G, Prusiner SB, Cohen FE. Folding of prion protein to its native alpha-helical conformation is under kinetic control. J Biol Chem 2001; 276:19687 - 19690
- Hafner-Bratkovic I, Bester R, Pristovsek P, Gaedtke L, Veranic P, Gaspersic J, et al. Globular domain of the prion protein needs to be unlocked by domain swapping to support prion protein conversion. J Biol Chem 2011; 286:12149 - 12156
- Koshland DE. Application of a Theory of Enzyme Specificity to Protein Synthesis. Proc Natl Acad Sci USA 1958; 44:98 - 104
- Wille H, Michelitsch MD, Guenebaut V, Supattapone S, Serban A, Cohen FE, et al. Structural studies of the scrapie prion protein by electron crystallography. Proc Natl Acad Sci USA 2002; 99:3563 - 3568
- Morrissey MP, Shakhnovich EI. Evidence for the role of PrP(C) helix 1 in the hydrophilic seeding of prion aggregates. Proc Natl Acad Sci USA 1999; 96:11293 - 11298
- Sawaya MR, Sambashivan S, Nelson R, Ivanova MI, Sievers SA, Apostol MI, et al. Atomic structures of amyloid cross-beta spines reveal varied steric zippers. Nature 2007; 447:453 - 457
- Vanik DL, Surewicz KA, Surewicz WK. Molecular basis of barriers for interspecies transmissibility of mammalian prions. Mol Cell 2004; 14:139 - 145
- Tateishi J, Kitamoto T, Hoque MZ, Furukawa H. Experimental transmission of Creutzfeldt-Jakob disease and related diseases to rodents. Neurology 1996; 46:532 - 537
- Khan MQ, Sweeting B, Mulligan VK, Arslan PE, Cashman NR, Pai EF, et al. Prion disease susceptibility is affected by beta-structure folding propensity and local side-chain interactions in PrP. Proc Natl Acad Sci USA 2010; 107:19808 - 19813
- Cobb NJ, Sonnichsen FD, McHaourab H, Surewicz WK. Molecular architecture of human prion protein amyloid: a parallel, in-register beta-structure. Proc Natl Acad Sci USA 2007; 104:18946 - 18951
- Cobb NJ, Surewicz WK. Prion strains under the magnifying glass. Nat Struct Mol Biol 2007; 14:882 - 884
- De Simone A, Zagari A, Derreumaux P. Structural and hydration properties of the partially unfolded states of the prion protein. Biophys J 2007; 93:1284 - 1292
- Chakroun N, Prigent S, Dreiss CA, Noinville S, Chapuis C, Fraternali F, et al. The oligomerization properties of prion protein are restricted to the H2H3 domain. FASEB J 2010; 24:3222 - 3231
- Adrover M, Pauwels K, Prigent S, de Chiara C, Xu Z, Chapuis C, et al. Prion fibrillization is mediated by a native structural element that comprises helices H2 and H3. J Biol Chem 2010; 285:21004 - 21012
- Lu X, Wintrode PL, Surewicz WK. Beta-sheet core of human prion protein amyloid fibrils as determined by hydrogen/deuterium exchange. Proc Natl Acad Sci USA 2007; 104:1510 - 1515
- Tycko R, Savtchenko R, Ostapchenko VG, Makarava N, Baskakov IV. The alpha-helical C-terminal domain of full-length recombinant PrP converts to an in-register parallel beta-sheet structure in PrP fibrils: Evidence from solid state nuclear magnetic resonance. Biochemistry 2010; 49:9488 - 9497
- Kryndushkin DS, Wickner RB, Tycko R. The core of Ure2p prion fibrils is formed by the N-terminal segment in a parallel cross-beta structure: Evidence from solid-state NMR. J Mol Biol 2011; 409:263 - 277
- Baskakov IV, Legname G, Baldwin MA, Prusiner SB, Cohen FE. Pathway complexity of prion protein assembly into amyloid. J Biol Chem 2002; 277:21140 - 21148
- Collinge J, Clarke AR. A general model of prion strains and their pathogenicity. Science 2007; 318:930 - 936
- Gorbenko GP, Kinnunen PK. The role of lipid-protein interactions in amyloid-type protein fibril formation. Chem Phys Lipids 2006; 141:72 - 82
- Morillas M, Swietnicki W, Gambetti P, Surewicz WK. Membrane environment alters the conformational structure of the recombinant human prion protein. J Biol Chem 1999; 274:36859 - 36865
- Wang F, Wang X, Yuan CG, Ma J. Generating a prion with bacterially expressed recombinant prion protein. Science 2010; 327:1132 - 1135
- Wang F, Yin S, Wang X, Zha L, Sy MS, Ma J. Role of the highly conserved middle region of prion protein (PrP) in PrP-lipid interaction. Biochemistry 2010; 49:8169 - 8176
- van der Kamp MW, Daggett V. The consequences of pathogenic mutations to the human prion protein. Protein Eng Des Sel 2009; 22:461 - 468
- Kuwata K, Nishida N, Matsumoto T, Kamatari YO, Hosokawa-Muto J, Kodama K, et al. Hot spots in prion protein for pathogenic conversion. Proc Natl Acad Sci USA 2007; 104:11921 - 11926
- Parchi P, Strammiello R, Giese A, Kretzschmar H. Phenotypic variability of sporadic human prion disease and its molecular basis: past, present and future. Acta Neuropathol 2011; 121:91 - 112
- Simoneau S, Rezaei H, Sales N, Kaiser-Schulz G, Lefebvre-Roque M, Vidal C, et al. In vitro and in vivo neurotoxicity of prion protein oligomers. PLoS Pathog 2007; 3:125
- Chich JF, Chapuis C, Henry C, Vidic J, Rezaei H, Noinville S. Vesicle permeabilization by purified soluble oligomers of prion protein: a comparative study of the interaction of oligomers and monomers with lipid membranes. J Mol Biol 2010; 397:1017 - 1030
- Noinville S, Chich JF, Rezaei H. Misfolding of the prion protein: linking biophysical and biological approaches. Vet Res 2008; 39:48