Abstract
Protein misfolding and assembly into ordered, self-templating aggregates (amyloid) has emerged as a novel mechanism for regulating protein function. For a subclass of amyloidogenic proteins known as prions, this process induces transmissible changes in normal cellular physiology, ranging from neurodegenerative disease in animals and humans to new traits in fungi. The severity and stability of these altered phenotypic states can be attenuated by the conformation or amino-acid sequence of the prion, but in most of these cases, the protein retains the ability to form amyloid in vitro. Thus, our ability to link amyloid formation in vitro with its biological consequences in vivo remains a challenge. In two recent studies, we have begun to address this disconnect by assessing the effects of the cellular environment on traits associated with the misfolding of the yeast prion Sup35. Remarkably, the effects of quality control pathways and of limitations on protein transfer in vivo amplify the effects of even slight differences in the efficiency of Sup35 misfolding, leading to dramatic changes in the associated phenotype. Together, our studies suggest that the interplay between protein misfolding pathways and their cellular context is a crucial contributor to prion biology.
Prion Propagation In Vivo
According to the prion hypothesis, an alternative conformation of a normal, host-encoded protein, known as a prion, can function as an epigenetic determinant of transmissible phenotypic states in vivo. This idea was originally proposed as a disease mechanism for the Transmissible Spongiform Encephalopathies (TSEs), a group of severe mammalian neurodegenerative disorders associated with the misfolding of the prion PrP.Citation1,Citation2 More recently, the prion hypothesis has provided a novel framework for understanding other enigmatic biological phenomena, including a group of metastable traits in fungi that are inherited through a non-Mendelian route.Citation3,Citation4 Although once met with skepticism, this protein-only mechanism has now been proven in multiple studies, where new transmissible phenotypes were induced in experimental organisms simply by introducing particular conformational variants (conformers) of prions.Citation5–Citation11
Prion-associated phenotypes are thought to arise because conformational conversion alters the activity of the protein.Citation4 However, this link between discrete physical and functional states of a protein only provides a steady-state snapshot of the endpoints of a dynamic process. The appearance, spread and reversal of prion-associated phenotypes necessarily involve changes in protein physical state.Citation12 However, these transitions are only effective if the alternative conformer is preferentially amplified, at the expense of all others, to a level that can alter cellular physiology.
Much of our understanding of this conformational selection and amplification results from studies in vitro, where the assembly of prion conformers into amyloid aggregates directs the continued misfolding of the protein by providing a template for this conversion.Citation13–Citation19 However, studies on the yeast prion Sup35 indicate that the process of conformational self-replication is more complex in vivo (, top), and a similar pathway has been proposed for prion propagation in mammals.Citation20 Based on this work, aggregates or their component prion-state protein must act in three roles to sustain the prion-associated phenotype in a population. As is the case in vitro, these aggregates template the conversion of newly made Sup35 to the prion conformer;Citation21,Citation22 however, in vivo, simple growth of the aggregates will not allow the spread of prion-associated phenotypes in a dividing culture. Thus, these aggregates also must be continually fragmented to create new conversion surfacesCitation23 and transmitted to daughter cells (, top).Citation23,Citation24
Prion Variants
While the pathway of prion propagation in vivo explains how the appearance of a misfolded conformer can be amplified to induce and maintain a new phenotype, this scheme alone is not sufficient to explain the diversity of prion-associated phenotypes. Remarkably, each prion has the capacity to create a spectrum of phenotypic states, known as strains or variants, which retain their characteristics upon serial passage in the same host.Citation25–Citation30 In mammals, variants of PrP are characterized by differences in host-specific incubation periods between initial infection and clinical disease,Citation31–Citation34 the distribution of pathological changes in the brain,Citation25,Citation33,Citation35 clinical symptoms,Citation36 sensitivity to thermal inactivationCitation37–Citation40 and the interspecies transmission of disease.Citation32,Citation38,Citation41–Citation43 In contrast, variants of the fungal prions do not specify new phenotypes but rather alter the severity of the prototypical prion-associated phenotype to generate a continuum of similar traits.Citation26,Citation27,Citation29
The existence of variants was once considered incompatible with a protein-only mechanism, but studies in both mammals and yeast have linked this phenotypic diversity to a parallel collection of unique conformers for each prion, which are characterized by changes in protease sensitivity, thermodynamic stability, and the extent of the core amyloid structure.Citation7,Citation25,Citation28,Citation44–Citation49 For both mammalian and fungal prions, these conformers are all self-replicated through the same pathway, but the physical differences between them affect the activities of aggregates and presumably their phenotypic consequences.Citation6,Citation7,Citation50 Aggregates composed of distinct prion conformers differ in their templating activityCitation49–Citation51 and in their thermodynamic stabilities, which likely affect both their ability to be fragmented and to be cleared by quality control pathways in vivo.Citation49,Citation52,Citation53 What has emerged from these analyses is that unique combinations of conversion and fragmentation efficiencies define prion variants, but how these biochemical parameters created distinct biological outcomes remained a major unanswered question.
Connecting Prion Misfolding to Phenotype
In general, the thermodynamic stability of mouse, both natural and synthetic (i.e., in vitro generated), and yeast prion aggregates is inversely correlated with the severity of their associated phenotypes (), most typically scored as incubation time in mammals and extent of aggregation in fungi.Citation49,Citation52,Citation53 Based on these observations, it was suggested that the efficiency of aggregate fragmentation in vivo, which should be a function of aggregate thermodynamic stability, has a profound effect on prion biology. If the system has reached steady-state, modest changes in fragmentation efficiency are predicted to inversely affect the size of aggregates and directly affect their accumulation, as prion synthesis is on-going,Citation54 and either (or both) of these variables could impact the creation of unique, conformation-based phenotypes. To distinguish between these size and abundance-based models of prion variants, we focused on the yeast protein Sup35, a component of the translation termination complex whose function is modulated by a prion cycle.Citation55–Citation57 In the non-prion [(psi−)]conformer, Sup35 is soluble and facilitates translation termination; however, in its prion [(PSI+)] conformers, Sup35 aggregates to different extents, establishing a corresponding range of decreased translation termination efficiencies.Citation21,Citation58,Citation59
Given the multiple roles of aggregates in prion propagation in vivo, we employed a computational approach to guide our experiments on the in vivo pathway through which prion variants are created. TSE propagation can be mathematically described in terms of PrP synthesis, conversion, fragmentation and decay rates,Citation20 and by substituting prion dilution during cell division for prion decay, the same formulation accurately captures the differences in soluble Sup35 levels found in two [PSI+] variants, known as weak and strong.Citation27,Citation49 However, this model cannot accurately predict other defining characteristics of yeast prion variants, including differences in the size distribution of aggregates,Citation60,Citation61 the spontaneous frequency of prion loss,Citation27,Citation62 and the elevated frequency of prion loss observed upon Sup35 overexpression,Citation63,Citation64 a phenomenon that is also observed for another yeast prion.Citation65–Citation67
Each of these characteristics is specific to prion propagation in vivo; therefore, we formulated a new model that incorporated aspects of the cellular environment that were known to impact prion propagation: the transmission of Sup35 protein to daughter cellsCitation23,Citation68 and the limitation on fragmentation efficiency imposed by the concentration of the molecular chaperone Hsp104, the catalyst for this reaction.Citation23,Citation69,Citation70 With this reformulation, we simulated [PSI+] propagation via a transmission model that was either based on aggregate abundance or size and assessed their accuracy in computationally recapitulating all of the characteristics of the [PSI+]weak and [PSI+]strong variants described above. In contrast to in vivo observations, the prion state was completely stable under all conditions in the abundance-based model because aggregates, if they accumulated to any extent, were efficiently transferred to daughter cells, as had been previously predicted.Citation62 However, the severities, stabilities and aggregate size distributions of [PSI+]weak and [PSI+]strong variants were accurately recapitulated if a size threshold for aggregate transmission was imposed, but only if this limitation fell within the size range of SDS-resistant aggregates that distinguished the two variants.Citation60
Our modeling suggested that the heritable prion species, known as a propagon,Citation71 represented a subset of aggregates that was distinguished simply by their size and that variants differed in their accumulation of these species. If true, the stability of variant-associated phenotypes should correlate with the proportion of Sup35 that was transmissible. To directly test this idea, we developed a fluorescence loss in photobleaching (FLIP) assay, in which daughter cells of yeast strains expressing Sup35-GFP are continually bleached prior to cytokinesis and fluorescence loss in the mother is monitored over time as a proxy for transmission (). Indeed, a [PSI+] weak variant transmitted 50% less Sup35-GFP and accumulated 50% fewer propagons than a [PSI+]strong variant, and these differences correlated with a three order of magnitude increase in prion loss during cell division (mitotic instability) for the [PSI+]weak variant.Citation27
If aggregate size alone is responsible for these effects, shifting this distribution, in the absence of conformational changes, should also impact both the transmissibility of Sup35 and the stability of the associated phenotype. In vivo, the steady-state size of aggregates is a function of both the conversion and fragmentation reactions; therefore, we varied Sup35 expression level to modulate the conversion rate. The size of SDS-resistant aggregates was increased by mild overexpression of Sup35 (∼5-fold) and dramatically decreased by repression of Sup35 synthesis for even one generation. By FLIP, conditions associated with the accumulation of larger aggregates resulted in a decrease in Sup35-GFP transmission (, middle), while those associated with smaller aggregates resulted in an increase in Sup35-GFP transmission. As was the case for different variants, these differences in transmission correlated directly with the number of propagons and indirectly with the mitotic stability of the prion-associated phenotype. Thus, our studies suggest that there is no inherent functional difference between aggregates of the same conformer but rather that their behavior in vivo is modulated by differential interactions with the cellular environment.
Conformation-dependent differences in prion-associated phenotypes were previously thought to arise from distinct equilibria between soluble and aggregated protein (i.e., abundance-based model) in all cells in a population.Citation49 However, our size-based model suggested that prion-associated phenotypes arose through a population-based mechanism. Specifically, the size threshold for aggregate transmission is predicted to create heterogeneity in the population, with mother cells accumulating more and larger aggregates than their daughters. By assessing Sup35 physical state in either mother or daughter cells by FLIP or a gel-based assay following centrifugal elutriation, we provided experimental support for this prediction. These age-dependent differences and the fact that prion variants produce phenotypically homogeneous colonies suggested that the aggregate complement and associated phenotype must change over time. Our modeling indeed predicted that the fraction of Sup35 in aggregates, which determines translation termination activity,Citation72 and their size both increased with replicative age. Consistent with this prediction, we demonstrated that the number of propagons transmitted to daughters increases with the replicative age of the mother and that the fidelity of translation termination, as monitored by a fluorescent reporter, decreases with replicative age. Thus, prion-associated phenotypes arise through a dynamic pathway of prion biogenesis rather than a simple equilibrium between the prion and non-prion conformers.
The size-dependent transmission of aggregates provides a framework for understanding prion-associated phenotypes in a dividing yeast culture, but can differences in aggregate size also provide insight into prion biology in mammalian post-mitotic neurons? Mathematical models have suggested that aggregate size as well as abundance could be important contributors to prion propagation in mammals.Citation20 Indeed, biochemical analyses have linked small aggregates to high infectivity and short incubation times.Citation73,Citation74 Mechanistically, aggregate size appears to impact converting activity,Citation73,Citation75 but others have suggested an effect on transmission as well.Citation20,Citation76 Consistent with this idea, more thermodynamically stable and presumably larger, aggregates are associated with localized pathology while less thermodynamically stable aggregates are associated with more widespread pathology.Citation77 Moreover, mammalian prion biology is altered by changes in expression of the prion protein, as is the case for yeast prion biology.Citation24,Citation63–Citation67 For example, the incubation time for clinical disease, but not for the generation of prion infectivity, is highly dependent on PrP expression level.Citation78,Citation79 While other mechanisms are possible,Citation78,Citation80,Citation81 an intriguing model to explain these observations is that infectivity increases with aggregate abundance until reaching a plateau, which represents a limitation on the system. If that limitation is fragmentation activity, aggregates will increase in size but not abundance, and this process would proceed more rapidly at higher PrP expression levels. In this scenario, prion toxicity, corresponding to clinical disease, could result from the titration of other cellular factors through their association with aggregates, a mechanism that has been proven for the toxicity of overexpressed Sup35 in [PSI+] yeast strains.Citation82 Thus, aggregate size may be an underappreciated contributor to the phenotypic outcomes of prion variants in mammals, as well.
Balancing Aggregate Assembly and Disassembly Pathways
While fragmentation efficiency appears to be a limitation on prion propagation in vivo, there may also be an upper boundary on the level of activity that is compatible with the prion state (). Several variants of hamster prion assemble into aggregates of lower thermodynamic stability than most other prion variants,Citation25,Citation32,Citation83,Citation84 but their incubation periods are significantly longer.Citation28,Citation49,Citation52 One potential explanation for these disconnects is that the rate of aggregate disassembly might approach that of aggregate assembly for these variants, leading to clearance.Citation50,Citation51,Citation84,Citation85 Consistent with this idea that aggregate assembly and disassembly pathways compete with one another in vivo, infectious particles delivered by direct inoculation, which presumably do not represent a load on the system, are largely cleared from experimental organisms,Citation78,Citation86–Citation90 and even established prion infections are reversed in vivo upon repression of new PrP synthesis.Citation91–Citation95
Other examples of the importance of competition between aggregate assembly and disassembly pathways in prion phenotypic outcomes may also be found in nature. Individuals with PrP polymorphisms are genetically less susceptible to prion disease.Citation96–Citation105 These polymorphisms clearly restrict the range of conformations accessible to the prion and therefore its ability to replicate certain variants.Citation31,Citation43,Citation106–Citation110 However, many of the same sequence changes also function as dominant inhibitors of prion propagation in vivo,Citation105,Citation111–Citation118 and a similar effect occurs upon co-expression of PrP homologues from different species.Citation79,Citation119–Citation124 Thus, these sequence variants must target crucial events in prion propagation by the wild-type protein, and elucidating their mechanisms of action could be instructive for developing therapeutic interventions for these diseases.Citation114–Citation116
Early models suggested that PrP dominant-negative mutants acted by titrating away a host-encoded cofactor (protein X) required for the conversion reaction;Citation122 however, these sequence variants also inhibit prion propagation in cell-free systems, suggesting that their effects are mediated directly through prion-prion interactions.Citation125–Citation127 In this scenario, PrP sequence variants would interact with the templating surface on an aggregate and either slow, as has been proposed for a Q219K variant, or block, as has been proposed for a Q172R variant, conversion of wild-type PrP, depending on their affinities for one another.Citation125,Citation126 Potentially supporting this affinity-based model, PrP dominant-negative mutants differ in their effective inhibitory ratios relative to wild-type protein,Citation111,Citation125–Citation129 but the same observation is also consistent with a model in which these mutants target different events in prion propagation in vivo. According to this latter idea, mutants that act at the templating surface would be effective at lower doses than those affecting fragmentation, which occurs along the length of the aggregate.Citation130
Propagation of the yeast prion [PSI+] is also disrupted by co-expression of Sup35 mutants, such as Q24R and G58D,Citation131–Citation133 and we used this system to explore the mechanisms underlying prion dominant inhibition in vivo.Citation134 We found that Q24R, like PrP Q172R, acted at sub-stochiometric ratios while G58D, like Q219K, interfered only at higher ratios relative to wild-type protein.Citation113,Citation125,Citation135 Using a series of in vivo analyses, we linked these differences to defects in discrete events in the prion propagation pathway. While we detected no defect in prion transmission by FLIP, we determined that G58D, but not Q24R, could efficiently join wild-type complexes by monitoring conversion by a translation termination defect that appears upon Sup35 incorporation into aggregates,Citation22 as had been previously suggested.Citation64,Citation131,Citation136,Citation137 In contrast, G58D, but not Q24R, incorporation into aggregates led to their destabilization, as measured by their sensitivity to disruption in SDS at increasing temperatures. Thus, Q24R expression decreases conversion efficiency while G58D expression likely increases fragmentation efficiency. In addition to revealing the molecular basis of the differences in effective inhibitory ratios, these effects on Sup35 biogenesis also explain the differential dominant interactions of Q24R and G58D with the [PSI+]weak variant, which is characterized by a reduced accumulation of aggregates due to inefficient fragmentation.Citation49 Q24R expression, which limits the conversion reaction and would further diminish aggregate accumulation, is completely incompatible with the [PSI+]weak variant (our unpublished observations), while G58D expression, which enhances fragmentation and would increase aggregate accumulation, promotes [PSI+]weak mitotic stability.Citation64
Consistent with these predictions, propagon accumulation in [PSI+]strong haploid strains decreased with Q24R expression but increased with G58D expression. However, these effects were modulated by the cellular environment: in [PSI+] strong diploid strains, expression of either mutant decreased propagon accumulation. We hypothesized that these observations could be explained by the relative doses of Sup35 and Hsp104 under the two conditions (2:1 vs. 1:1, respectively). If true, modulating chaperone levels in diploids should affect the severity of the inhibition. Indeed, the dominant-negative effects of both Q24R and G58D were partially or completely reversed, respectively, by lowering Hsp104 levels and were enhanced by overexpression of Hsp104. Moreover, we found differences in the Sup35:Hsp104 ratio in yeast strains of different genetic backgrounds, providing a molecular explanation for known variations in G58D efficacy.Citation64,Citation132
The dependency of prion inhibition on chaperone level suggested that the disassembly pathway was competing more efficiently with the assembly pathway in the presence of the dominant-negative mutants. To determine if aggregates were actually being resolubilized, we monitored the fate of existing Sup35 after treating [PSI+]strong cultures with cycloheximide. In these experiments, Sup35 transitioned from an SDS-resistant to an SDS-sensitive form in yeast strains expressing both wild-type and dominant-negative mutants but not in those expressing wild-type protein alone. Mechanistically, we predict that Q24R and G58D induce this same outcome through different pathways: the conversion defect of Q24R would increase the ratio of enzyme (Hsp104) to substrate (aggregates) for the fragmentation reaction, while the efficiency of that reaction would increase in the presence of G58D (, bottom). Thus, in either case, expression of dominant-negative mutants skews the competition between aggregate assembly and disassembly pathways toward the latter, allowing the effective clearance of an established prion conformer in vivo (). Given the heterogeneity in aggregate accumulation that we have observed in individual wild-type yeast cells, the balance between these opposing forces may also vary among cells in the population, contributing a currently unappreciated influence on the severity and stability of prion-associated phenotypes under normal conditions.
Strikingly, there are many parallels between Sup35 Q24R and PrP Q172R and between Sup35 G58D and PrP Q219K. In addition to displaying similar effective inhibitory ratios, PrP Q172R, like Sup35 Q24R, is conversion defective and is incompatible with prion variants of different thermodynamic stabilities.Citation28,Citation117,Citation125,Citation135 In contrast, PrP Q219K, like, Sup35 G58D, converts to the prion conformer efficiently, destabilizes aggregates and is incompatible only with prion variants of reduced thermodynamic stability.Citation28,Citation105,Citation117,Citation125,Citation126 Thus, despite the distinct cellular environments in which mammalian and yeast prions propagate, the pathways of dominant inhibition that we have uncovered for Sup35 mutants may be applicable to PrP mutants and may provide mechanistic insight into the phenotypic outcomes of prion infections in that system.
Conclusions
Despite our detailed understanding of amyloidogenesis in vitro, our ability to mechanistically link this process to its biological consequences in vivo lags far behind.Citation138,Citation139 Collectively, our studies on the yeast prion [PSI+] have begun to bridge this gap by demonstrating that the cellular environment dramatically influences the physiological consequences of Sup35 misfolding. Notably, conformation-based phenotypes are created through the interplay among Sup35 folding, protein quality control and other aspects of yeast cell biology,Citation24 and imbalances in these forces lead to the dominant inhibition of prion propagation in vivo.Citation134 Given the many parallels between prion propagation in yeast and in mammals, the insight gained through these studies may provide a new framework for connecting protein misfolding pathways in vitro to disease mechanisms in vivo.
Note Added in Proof
Recent studies by the Weissman group have determined that amyloid fibers composed of G58D alone, when present in a particular conformation, exhibit the same conversion and thermodynamic stability differences in comparison with wild-type fibers in vitro as we have identified for mixed G58D-wild-type prion aggregates in vivo.Citation140
Figures and Tables
Figure 1 The cellular environment modulates prion misfolding pathways to create protein-based traits. (top) Self-replicating protein conformations create stable phenotypes (white colonies) in vivo when the processes of synthesis (1), conversion (2), fragmentation (3) by Hsp104 (hexamer) and transmission (4) are balanced to allow aggregates of prion proteins to persist in vivo. (middle) Overexpression of a prion protein promotes the conversion reaction (red arrows), leading to the accumulation of large aggregates that are inefficiently transmitted to daughter cells (dotted red arrow) and loss of the prion-associated phenotype (red colonies). (bottom) Dominant inhibition of prion propagation by mutants that decrease conversion efficiency (dotted red arrow) or enhance fragmentation efficiency (solid red arrow) promote aggregate disassembly (ball and stick) and induce prion loss (red colonies).
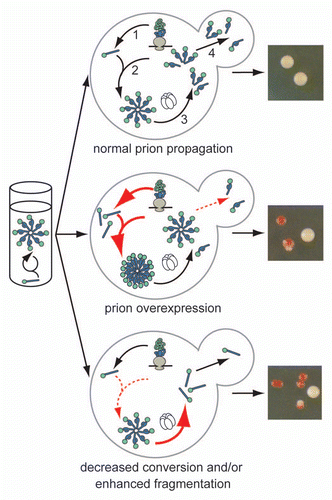
Figure 2 Relating prion phenotypic severity to aggregate thermodynamic stability. For many prion variants, there is a linear but inverse relationship between aggregate thermodynamic stability and phenotypic severity (black dotted line), but this trend cannot explain the phenotypes associated with all prion variants or the effects of dominant-negative prion mutants (see text for details). Our studies in vivo on the yeast prion [PSI+] suggest that thermodynamic stability poses a limit on prion persistence at both extremes (red line) by impacting aggregate size and accumulation (shown schematically). The least thermodynamically stable aggregates are efficiently resolubilized, while the most thermodynamically stable aggregates are inefficiently transmitted.
![Figure 2 Relating prion phenotypic severity to aggregate thermodynamic stability. For many prion variants, there is a linear but inverse relationship between aggregate thermodynamic stability and phenotypic severity (black dotted line), but this trend cannot explain the phenotypes associated with all prion variants or the effects of dominant-negative prion mutants (see text for details). Our studies in vivo on the yeast prion [PSI+] suggest that thermodynamic stability poses a limit on prion persistence at both extremes (red line) by impacting aggregate size and accumulation (shown schematically). The least thermodynamically stable aggregates are efficiently resolubilized, while the most thermodynamically stable aggregates are inefficiently transmitted.](/cms/asset/56823a06-0c44-444e-9b76-72fec2b5b72a/kprn_a_10916413_f0002.gif)
Figure 3 Transmission of Sup35 protein to daughter cells is conformation-dependent. (left) Schematic of fluorescence loss in photobleaching assay (FLIP) for Sup35 transmission to daughter cells. Bleached daughter (red) and monitored mother (black) are indicated. (right) Fluorescence retention in mother cells expressing Sup35-GFP in the [PSI+]strong (white), [PSI+]weak (pink) or [psi−] (red) conformation.
![Figure 3 Transmission of Sup35 protein to daughter cells is conformation-dependent. (left) Schematic of fluorescence loss in photobleaching assay (FLIP) for Sup35 transmission to daughter cells. Bleached daughter (red) and monitored mother (black) are indicated. (right) Fluorescence retention in mother cells expressing Sup35-GFP in the [PSI+]strong (white), [PSI+]weak (pink) or [psi−] (red) conformation.](/cms/asset/5ef91a15-3bc9-44c0-b8c9-92c0bc548768/kprn_a_10916413_f0003.gif)
Acknowledgments
We thank A. Derdowski for the FLIP image and members of the Serio lab for helpful discussions and comments on the manuscript. This work was supported by awards from the National Institutes of Health to T.R.S. (GM069802) and to S.D. (AG032818).
References
- Griffith JS. Self-replication and scrapie. Nature 1967; 215:1043 - 1044
- Prusiner SB. Novel proteinaceous infectious particles cause scrapie. Science 1982; 216:136 - 144
- Wickner RB. [URE3] as an altered URE2 protein: evidence for a prion analog in Saccharomyces cerevisiae. Science 1994; 264:566 - 569
- Tuite MF, Serio TR. The prion hypothesis: from biological anomaly to basic regulatory mechanism. Nat Rev Mol Cell Biol 2010; 11:823 - 833
- Brachmann A, Baxa U, Wickner RB. Prion generation in vitro: amyloid of Ure2p is infectious. EMBO J 2005; 24:3082 - 3092
- King CY, Diaz-Avalos R. Protein-only transmission of three yeast prion strains. Nature 2004; 428:319 - 323
- Tanaka M, Chien P, Naber N, Cooke R, Weissman JS. Conformational variations in an infectious protein determine prion strain differences. Nature 2004; 428:323 - 328
- Patel BK, Liebman SW. “Prion-proof” for [PIN+]: Infection with in vitro-made amyloid aggregates of Rnq1p-(132–405) induces [PIN+]. J Mol Biol 2007; 365:773 - 782
- Maddelein ML, Dos Reis S, Duvezin-Caubet S, Coulary-Salin B, Saupe SJ. Amyloid aggregates of the HET-s prion protein are infectious. Proc Natl Acad Sci USA 2002; 99:7402 - 7407
- Kim JI, Cali I, Surewicz K, Kong Q, Raymond GJ, Atarashi R, et al. Mammalian prions generated from bacterially expressed prion protein in the absence of any mammalian cofactors. J Biol Chem 2010; 285:14083 - 14087
- Wang F, Wang X, Yuan CG, Ma J. Generating a prion with bacterially expressed recombinant prion protein. Science 2010; 327:1132 - 1135
- Pezza JA, Serio TR. Prion propagation: The role of protein dynamics. Prion 2007; 1:36 - 43
- Sondheimer N, Lindquist S. Rnq1: an epigenetic modifier of protein function in yeast. Mol Cell 2000; 5:163 - 172
- Kocisko DA, Come JH, Priola SA, Chesebro B, Raymond GJ, Lansbury PT, et al. Cell-free formation of protease-resistant prion protein. Nature 1994; 370:471 - 474
- Glover JR, Kowal AS, Schirmer EC, Patino MM, Liu JJ, Lindquist S. Self-seeded fibers formed by Sup35, the protein determinant of [PSI+], a heritable prion-like factor of S. cerevisiae. Cell 1997; 89:811 - 819
- King CY, Tittmann P, Gross H, Gebert R, Aebi M, Wuthrich K. Prion-inducing domain 2–114 of yeast Sup35 protein transforms in vitro into amyloid-like filaments. Proc Natl Acad Sci USA 1997; 94:6618 - 6622
- Taylor KL, Cheng N, Williams RW, Steven AC, Wickner RB. Prion domain initiation of amyloid formation in vitro from native Ure2p. Science 1999; 283:1339 - 1343
- Saborio GP, Permanne B, Soto C. Sensitive detection of pathological prion protein by cyclic amplification of protein misfolding. Nature 2001; 411:810 - 813
- Dos Reis S, Coulary-Salin B, Forge V, Lascu I, Begueret J, Saupe SJ. The HET-s prion protein of the filamentous fungus Podospora anserina aggregates in vitro into amyloid-like fibrils. J Biol Chem 2002; 277:5703 - 5706
- Masel J, Jansen VA, Nowak MA. Quantifying the kinetic parameters of prion replication. Biophys Chem 1999; 77:139 - 152
- Patino MM, Liu JJ, Glover JR, Lindquist S. Support for the prion hypothesis for inheritance of a phenotypic trait in yeast. Science 1996; 273:622 - 626
- Satpute-Krishnan P, Serio TR. Prion protein remodelling confers an immediate phenotypic switch. Nature 2005; 437:262 - 265
- Satpute-Krishnan P, Langseth SX, Serio TR. Hsp104-dependent remodeling of prion complexes mediates protein-only inheritance. PLoS Biol 2007; 5:24
- Derdowski A, Sindi SS, Klaips CL, DiSalvo S, Serio TR. A size threshold limits prion transmission and establishes phenotypic diversity. Science 2010; 330:680 - 683
- Bessen RA, Marsh RF. Distinct PrP properties suggest the molecular basis of strain variation in transmissible mink encephalopathy. J Virol 1994; 68:7859 - 7868
- Bradley ME, Edskes HK, Hong JY, Wickner RB, Liebman SW. Interactions among prions and prion “strains” in yeast. Proc Natl Acad Sci USA 2002; 99:16392 - 16399
- Derkatch IL, Chernoff YO, Kushnirov VV, Inge-Vechtomov SG, Liebman SW. Genesis and variability of [PSI] prion factors in Saccharomyces cerevisiae. Genetics 1996; 144:1375 - 1386
- Safar J, Wille H, Itri V, Groth D, Serban H, Torchia M, et al. Eight prion strains have PrP(Sc) molecules with different conformations. Nat Med 1998; 4:1157 - 1165
- Schlumpberger M, Prusiner SB, Herskowitz I. Induction of distinct [URE3] yeast prion strains. Mol Cell Biol 2001; 21:7035 - 7046
- Wickner RB, Edskes HK, Shewmaker F, Kryndushkin D, Nemecek J. Prion variants, species barriers, generation and propagation. J Biol 2009; 8:47
- Dickinson AG, Meikle VM. Host-genotype and agent effects in scrapie incubation: change in allelic interaction with different strains of agent. Mol Gen Genet 1971; 112:73 - 79
- Bessen RA, Marsh RF. Identification of two biologically distinct strains of transmissible mink encephalopathy in hamsters. J Gen Virol 1992; 73:329 - 334
- Bruce ME, McConnell I, Fraser H, Dickinson AG. The disease characteristics of different strains of scrapie in Sinc congenic mouse lines: implications for the nature of the agent and host control of pathogenesis. J Gen Virol 1991; 72:595 - 603
- Pattison IH, Millson GC. Scrapie produced experimentally in goats with special reference to the clinical syndrome. J Comp Path 1961; 71:101 - 109
- Fraser H, Dickinson AG. Scrapie in mice. Agent-strain differences in the distribution and intensity of grey matter vacuolation. J Comp Path 1973; 83:29 - 40
- Carp RI, Callahan SM, Sersen EA, Moretz RC. Preclinical changes in weight of scrapie-infected mice as a function of scrapie agent-mouse strain combination. Intervirology 1984; 21:61 - 69
- Dickinson AG, Taylor DM. Resistance of scrapie agent to decontamination. N Engl J Med 1978; 299:1413 - 1414
- Kimberlin RH, Walker CA. Evidence that the transmission of one source of scrapie agent to hamsters involves separation of agent strains from a mixture. J Gen Virol 1978; 39:487 - 496
- Kimberlin RH, Walker CA, Millson GC, Taylor DM, Robertson PA, Tomlinson AH, et al. Disinfection studies with two strains of mouse-passaged scrapie agent. Guidelines for Creutzfeldt-Jakob and related agents. J Neurol Sci 1983; 59:355 - 369
- Taylor DM, Fernie K, McConnell I, Steele PJ. Observations on thermostable subpopulations of the unconventional agents that cause transmissible degenerative encephalopathies. Vet Microbiol 1998; 64:33 - 38
- Kimberlin RH, Cole S, Walker CA. Temporary and permanent modifications to a single strain of mouse scrapie on transmission to rats and hamsters. J Gen Biol 1987; 68:1875 - 1881
- Asante EA, Linehan JM, Desbruslais M, Joiner S, Gowland I, Wood AL, et al. BSE prions propagate as either variant CJD-like or sporadic CJD-like prion strains in transgenic mice expressing human prion protein. EMBO J 2002; 21:6358 - 6366
- Wadsworth JD, Asante EA, Desbruslais M, Linehan JM, Joiner S, Gowland I, et al. Human prion protein with valine 129 prevents expression of variant CJD phenotype. Science 2004; 306:1793 - 1796
- Caughey B, Raymond GJ, Bessen RA. Strain-dependent differences in beta-sheet conformations of abnormal prion protein. J Biol Chem 1998; 273:32230 - 32235
- Toyama BH, Kelly MJ, Gross JD, Weissman JS. The structural basis of yeast prion strain variants. Nature 2007; 449:233 - 237
- Peretz D, Williamson RA, Legname G, Matsunaga Y, Vergara J, Burton DR, et al. A change in the conformation of prions accompanies the emergence of a new prion strain. Neuron 2002; 34:921 - 932
- Telling GC, Parchi P, DeArmond SJ, Cortelli P, Montagna P, Gabizon R, et al. Evidence for the conformation of the pathologic isoform of the prion protein enciphering and propagating prion diversity. Science 1996; 274:2079 - 2082
- Peretz D, Scott MR, Groth D, Williamson RA, Burton DR, Cohen FE, et al. Strain-specified relative conformational stability of the scrapie prion protein. Protein Sci 2001; 10:854 - 863
- Tanaka M, Collins SR, Toyama BH, Weissman JS. The physical basis of how prion conformations determine strain phenotypes. Nature 2006; 442:585 - 589
- Bessen RA, Kocisko DA, Raymond GJ, Nandan S, Lansbury PT, Caughey B. Non-genetic propagation of strain-specific properties of scrapie prion protein. Nature 1995; 375:698 - 700
- Mulcahy ER, Bessen RA. Strain-specific kinetics of prion protein formation in vitro and in vivo. J Biol Chem 2004; 279:1643 - 1649
- Legname G, Nguyen HO, Peretz D, Cohen FE, DeArmond SJ, Prusiner SB. Continuum of prion protein structures enciphers a multitude of prion isolate-specified phenotypes. Proc Natl Acad Sci USA 2006; 103:19105 - 19110
- Prusiner SB, Scott MR, DeArmond SJ, Cohen FE. Prion protein biology. Cell 1998; 93:337 - 348
- Sindi SS, Serio TR. Prion dynamics and the quest for the genetic determinant in protein-only inheritance. Curr Opin Microbiol 2009; 12:623 - 630
- Serio TR, Lindquist SL. [PSI+]: an epigenetic modulator of translation termination efficiency. Annu Rev Cell Dev Biol 1999; 15:661 - 703
- Stansfield I, Jones KM, Kushnirov VV, Dagkesamanskaya AR, Poznyakovski AI, Paushkin SV, et al. The products of the SUP45 (eRF1) and SUP35 genes interact to mediate translation termination in Saccharomyces cerevisiae. EMBO J 1995; 14:4365 - 4373
- Zhouravleva G, Frolova L, Le Goff X, Le Guellec R, Inge-Vechtomov S, Kisselev L, et al. Termination of translation in eukaryotes is governed by two interacting polypeptide chain release factors, eRF1 and eRF3. EMBO J 1995; 14:4065 - 4072
- Cox B. [PSI], a cytoplasmic suppressor of super-suppression in yeast. Heredity 1965; 20:505 - 521
- Paushkin SV, Kushnirov VV, Smirnov VN, Ter-Avanesyan MD. Propagation of the yeast prion-like [psi+] determinant is mediated by oligomerization of the SUP35-encoded polypeptide chain release factor. EMBO J 1996; 15:3127 - 3134
- Kryndushkin DS, Alexandrov IM, Ter-Avanesyan MD, Kushnirov VV. Yeast [PSI+] prion aggregates are formed by small Sup35 polymers fragmented by Hsp104. J Biol Chem 2003; 278:49636 - 49643
- Bagriantsev SN, Kushnirov VV, Liebman SW. Analysis of amyloid aggregates using agarose gel electrophoresis. Meth Enz 2006; 412:33 - 48
- Byrne LJ, Cole DJ, Cox BS, Ridout MS, Morgan BJ, Tuite MF. The number and transmission of [PSI] prion seeds (Propagons) in the yeast Saccharomyces cerevisiae. PLoS One 2009; 4:4670
- Allen KD, Wegrzyn RD, Chernova TA, Muller S, Newnam GP, Winslett PA, et al. Hsp70 chaperones as modulators of prion life cycle: novel effects of Ssa and Ssb on the Saccharomyces cerevisiae prion [PSI+]. Genetics 2005; 169:1227 - 1242
- Derkatch IL, Bradley ME, Zhou P, Liebman SW. The PNM2 mutation in the prion protein domain of SUP35 has distinct effects on different variants of the [PSI+] prion in yeast. Curr Genet 1999; 35:59 - 67
- Crapeau M, Marchal C, Cullin C, Maillet L. The cellular concentration of the yeast Ure2p prion protein affects its propagation as a prion. Mol Biol Cell 2009; 20:2286 - 2296
- Edskes HK, Gray VT, Wickner RB. The [URE3] prion is an aggregated form of Ure2p that can be cured by overexpression of Ure2p fragments. Proc Natl Acad Sci USA 1999; 96:1498 - 1503
- Baudin-Baillieu A, Fernandez-Bellot E, Reine F, Coissac E, Cullin C. Conservation of the prion properties of Ure2p through evolution. Mol Biol Cell 2003; 14:3449 - 3458
- Kawai-Noma S, Pack CG, Tsuji T, Kinjo M, Taguchi H. Single mother-daughter pair analysis to clarify the diffusion properties of yeast prion Sup35 in guanidine-HCl-treated [PSI] cells. Genes Cells 2009; 4:1045 - 1054
- Haslberger T, Bukau B, Mogk A. Towards a unifying mechanism for ClpB/Hsp104-mediated protein disaggregation and prion propagation. Biochem Cell Biol 2010; 88:63 - 75
- Ness F, Ferreira P, Cox BS, Tuite MF. Guanidine hydrochloride inhibits the generation of prion “seeds” but not prion protein aggregation in yeast. Mol Cell Biol 2002; 22:5593 - 5605
- Cox B, Ness F, Tuite M. Analysis of the generation and segregation of propagons: entities that propagate the [PSI+] prion in yeast. Genetics 2003; 165:23 - 33
- Zhou P, Derkatch IL, Uptain SM, Patino MM, Lindquist S, Liebman SW. The yeast non-Mendelian factor [ETA+] is a variant of [PSI+], a prion-like form of release factor eRF3. EMBO J 1999; 18:1182 - 1191
- Silveira JR, Raymond GJ, Hughson AG, Race RE, Sim VL, Hayes SF, et al. The most infectious prion protein particles. Nature 2005; 437:257 - 261
- Tixador P, Herzog L, Reine F, Jaumain E, Chapuis J, Le Dur A, et al. The physical relationship between infectivity and prion protein aggregates is strain-dependent. PLoS Path 2010; 6:1000859
- Calvez V, Lenuzza N, Oelz D, Deslys JP, Laurent P, Mouthon F, et al. Size distribution dependence of prion aggregates infectivity. Math Biosci 2009; 217:88 - 99
- Zampieri M, Legname G, Altafini C. Investigating the conformational stability of prion strains through a kinetic replication model. PLoS Comput Biol 2009; 5:1000420
- Legname G, Nguyen HO, Baskakov IV, Cohen FE, Dearmond SJ, Prusiner SB. Strain-specified characteristics of mouse synthetic prions. Proc Natl Acad Sci USA 2005; 102:2168 - 2173
- Sandberg MK, Al-Doujaily H, Sharps B, Clarke AR, Collinge J. Prion propagation and toxicity in vivo occur in two distinct mechanistic phases. Nature 2011; 470:540 - 542
- Scott M, Foster D, Mirenda C, Serban D, Coufal F, Walchli M, et al. Transgenic mice expressing hamster prion protein produce species-specific scrapie infectivity and amyloid plaques. Cell 1989; 59:847 - 857
- Wickner RB. Prion diseases: Infectivity versus toxicity. Nature 2011; 470:470 - 471
- Ghaemmaghami S, Phuan PW, Perkins B, Ullman J, May BC, Cohen FE, et al. Cell division modulates prion accumulation in cultured cells. Proc Natl Acad Sci USA 2007; 104:17971 - 17976
- Vishveshwara N, Bradley ME, Liebman SW. Sequestration of essential proteins causes prion associated toxicity in yeast. Mol Microbiol 2009; 73:1101 - 1114
- Bessen RA, Marsh RF. Biochemical and physical properties of the prion protein from two strains of the transmissible mink encephalopathy agent. J Virol 1992; 66:2096 - 2101
- Ayers JI, Schutt CR, Shikiya RA, Aguzzi A, Kincaid AE, Bartz JC. The strain-encoded relationship between PrP replication, stability and processing in neurons is predictive of the incubation period of disease. PLoS Path 2011; 7:1001317
- Shikiya RA, Ayers JI, Schutt CR, Kincaid AE, Bartz JC. Coinfecting prion strains compete for a limiting cellular resource. J Virol 2010; 84:5706 - 5714
- Bueler H, Aguzzi A, Sailer A, Greiner RA, Autenried P, Aguet M, et al. Mice devoid of PrP are resistant to scrapie. Cell 1993; 73:1339 - 1347
- Dickinson AG, Fraser H, Outram GW. Scrapie incubation time can exceed natural lifespan. Nature 1975; 256:732 - 733
- Race R, Raines A, Raymond GJ, Caughey B, Chesebro B. Long-term subclinical carrier state precedes scrapie replication and adaptation in a resistant species: analogies to bovine spongiform encephalopathy and variant Creutzfeldt-Jakob disease in humans. J Virol 2001; 75:10106 - 10112
- Safar JG, Kellings K, Serban A, Groth D, Cleaver JE, Prusiner SB, et al. Search for a prion-specific nucleic acid. J Virol 2005; 79:10796 - 10806
- Outram GW. Kimberlin RH. The pathogenesis of scrapie in mice. Slow Virus Diseases of Animals and Man 1976; Amsterdam North-Holland 325 - 357
- Safar JG, DeArmond SJ, Kociuba K, Deering C, Didorenko S, Bouzamondo-Bernstein E, et al. Prion clearance in bigenic mice. J Gen Virol 2005; 86:2913 - 2923
- Mallucci G, Dickinson A, Linehan J, Klohn PC, Brandner S, Collinge J. Depleting neuronal PrP in prion infection prevents disease and reverses spongiosis. Science 2003; 302:871 - 874
- Mallucci GR, White MD, Farmer M, Dickinson A, Khatun H, Powell AD, et al. Targeting cellular prion protein reverses early cognitive deficits and neurophysiological dysfunction in prion-infected mice. Neuron 2007; 53:325 - 335
- Pfeifer A, Eigenbrod S, Al-Khadra S, Hofmann A, Mitteregger G, Moser M, et al. Lentivector-mediated RNAi efficiently suppresses prion protein and prolongs survival of scrapie-infected mice. J Clin Invest 2006; 116:3204 - 3210
- White MD, Farmer M, Mirabile I, Brandner S, Collinge J, Mallucci GR. Single treatment with RNAi against prion protein rescues early neuronal dysfunction and prolongs survival in mice with prion disease. Proc Natl Acad Sci USA 2008; 105:10238 - 10243
- Goldmann W, Chong A, Foster J, Hope J, Hunter N. The shortest known prion protein gene allele occurs in goats, has only three octapeptide repeats and is non-pathogenic. J Gen Virol 1998; 79:3173 - 3176
- Dickinson AG, Stamp JT, Renwick CC, Rennie JC. Some factors controlling the incidence of scrapie in Cheviot sheep injected with a Cheviot-passaged scrapie agent. J Comp Path 1968; 78:313 - 321
- Hunter N, Moore L, Hosie BD, Dingwall WS, Greig A. Association between natural scrapie and PrP genotype in a flock of Suffolk sheep in Scotland. Vet Rec 1997; 140:59 - 63
- Laplanche JL, Delasnerie-Laupretre N, Brandel JP, Chatelain J, Beaudry P, Alperovitch A, et al. Molecular genetics of prion diseases in France. French research group on epidemiology of human spongiform encephalopathies. Neurology 1994; 44:2347 - 2351
- Belt PB, Muileman IH, Schreuder BE, Bos-de Ruijter J, Gielkens AL, Smits MA. Identification of five allelic variants of the sheep PrP gene and their association with natural scrapie. J Gen Virol 1995; 76:509 - 517
- Maciulis A, Hunter N, Wang S, Goldmann W, Hope J, Foote WC. Polymorphisms of a scrapie-associated fibril protein (PrP) gene and their association with susceptibility to experimentally induced scrapie in Cheviot sheep in the United States. Am J Vet Res 1992; 53:1957 - 1960
- Dickenson AG, Outram GW. Genetic aspects of unconventional virus infections: the basis fo the virino hypothesis. Ciba Foundation Symposium 1988; 135:63 - 83
- Bossers A, de Vries R, Smits MA. Susceptibility of sheep for scrapie as assessed by in vitro conversion of nine naturally occurring variants of PrP. J Virol 2000; 74:1407 - 1414
- Shibuya S, Higuchi J, Shin RW, Tateishi J, Kitamoto T. Codon 219 Lys allele of PRNP is not found in sporadic Creutzfeldt-Jakob disease. Ann Neurol 1998; 43:826 - 828
- Hizume M, Kobayashi A, Teruya K, Ohashi H, Ironside JW, Mohri S, et al. Human prion protein (PrP) 219K is converted to PrPSc but shows heterozygous inhibition in variant Creutzfeldt-Jakob disease infection. J Biol Chem 2009; 284:3603 - 3609
- Parchi P, Castellani R, Capellari S, Ghetti B, Young K, Chen SG, et al. Molecular basis of phenotypic variability in sporadic Creutzfeldt-Jakob disease. Ann Neurol 1996; 39:767 - 778
- Parchi P, Giese A, Capellari S, Brown P, Schulz-Schaeffer W, Windl O, et al. Classification of sporadic Creutzfeldt-Jakob disease based on molecular and phenotypic analysis of 300 subjects. Ann Neurol 1999; 46:224 - 233
- Collinge J, Sidle KC, Meads J, Ironside J, Hill AF. Molecular analysis of prion strain variation and the aetiology of ‘new variant’ CJD. Nature 1996; 383:685 - 690
- Hill AF, Joiner S, Wadsworth JD, Sidle KC, Bell JE, Budka H, et al. Molecular classification of sporadic Creutzfeldt-Jakob disease. Brain 2003; 126:1333 - 1346
- Collinge J, Clarke AR. A general model of prion strains and their pathogenicity. Science 2007; 318:930 - 936
- Kaneko K, Zulianello L, Scott M, Cooper CM, Wallace AC, James TL, et al. Evidence for protein X binding to a discontinuous epitope on the cellular prion protein during scrapie prion propagation. Proc Natl Acad Sci USA 1997; 94:10069 - 10074
- Zulianello L, Kaneko K, Scott M, Erpel S, Han D, Cohen FE, et al. Dominant-negative inhibition of prion formation diminished by deletion mutagenesis of the prion protein. J Virol 2000; 74:4351 - 4360
- Perrier V, Kaneko K, Safar J, Vergara J, Tremblay P, DeArmond SJ, et al. Dominant-negative inhibition of prion replication in transgenic mice. Proc Natl Acad Sci USA 2002; 99:13079 - 13084
- Crozet C, Lin Y, Mettling C, Mourton-Gilles C, Corbeau P, Lehmann S, et al. Inhibition of PrPSc formation by lentiviral gene transfer of PrP containing dominant negative mutants. J Cell Sci 2004; 117:5591 - 5597
- Furuya K, Kawahara N, Yamakawa Y, Kishida H, Hachiya NS, Nishijima M, et al. Intracerebroventricular delivery of dominant negative prion protein in a mouse model of iatrogenic Creutzfeldt-Jakob disease after dura graft transplantation. Neurosci Lett 2006; 402:222 - 226
- Toupet K, Compan V, Crozet C, Mourton-Gilles C, Mestre-Frances N, Ibos F, et al. Effective gene therapy in a mouse model of prion diseases. PLoS One 2008; 3:2773
- Atarashi R, Sim VL, Nishida N, Caughey B, Katamine S. Prion strain-dependent differences in conversion of mutant prion proteins in cell culture. J Virol 2006; 80:7854 - 7862
- Ott D, Taraborrelli C, Aguzzi A. Novel dominant-negative prion protein mutants identified from a randomized library. Protein Eng Des Sel 2008; 21:623 - 629
- Prusiner S, Scott M, Foster D, Pan K, Groth D, Mirenda C, et al. Transgenetic studies implicate interactions between homologous PrP isoforms in scrapie prion replication. Cell 1990; 63:673 - 686
- Scott MR, Kohler R, Foster D, Prusiner SB. Chimeric prion protein expression in cultured cells and transgenic mice. Protein Sci 1992; 1:986 - 997
- Prusiner SB, Groth D, Serban A, Koehler R, Foster D, Torchia M, et al. Ablation of the prion protein (PrP) gene in mice prevents scrapie and facilitates production of anti-PrP antibodies. Proc Natl Acad Sci USA 1993; 90:10608 - 10612
- Telling GC, Scott M, Mastrianni J, Gabizon R, Torchia M, Cohen FE, et al. Prion propagation in mice expressing human and chimeric PrP transgenes implicates the interaction of cellular PrP with another protein. Cell 1995; 83:79 - 90
- Telling GC, Scott M, Hsiao KK, Foster D, Yang SL, Torchia M, et al. Transmission of Creutzfeldt-Jakob disease from humans to transgenic mice expressing chimeric human-mouse prion protein. Proc Natl Acad Sci USA 1994; 91:9936 - 9940
- Korth C, Kaneko K, Groth D, Heye N, Telling G, Mastrianni J, et al. Abbreviated incubation times for human prions in mice expressing a chimeric mouse-human prion protein transgene. Proc Natl Acad Sci USA 2003; 100:4784 - 4789
- Geoghegan JC, Miller MB, Kwak AH, Harris BT, Supattapone S. Trans-dominant inhibition of prion propagation in vitro is not mediated by an accessory cofactor. PLoS Path 2009; 5:1000535
- Lee CI, Yang Q, Perrier V, Baskakov IV. The dominant-negative effect of the Q218K variant of the prion protein does not require protein X. Protein Sci 2007; 16:2166 - 2173
- Horiuchi M, Priola SA, Chabry J, Caughey B. Interactions between heterologous forms of prion protein: binding, inhibition of conversion and species barriers. Proc Natl Acad Sci USA 2000; 97:5836 - 5841
- Priola SA. Prion protein and species barriers in the transmissible spongiform encephalopathies. Biomed Pharmacother 1999; 53:27 - 33
- Priola SA, Caughey B, Race RE, Chesebro B. Heterologous PrP molecules interfere with accumulation of protease-resistant PrP in scrapie-infected murine neuroblastoma cells. J Virol 1994; 68:4873 - 4878
- Masel J, Jansen VA. Designing drugs to stop the formation of prion aggregates and other amyloids. Biophys Chem 2000; 88:47 - 59
- DePace AH, Santoso A, Hillner P, Weissman JS. A critical role for amino-terminal glutamine/asparagine repeats in the formation and propagation of a yeast prion. Cell 1998; 93:1241 - 1252
- Doel SM, McCready SJ, Nierras CR, Cox BS. The dominant PNM2-mutation which eliminates the psi factor of Saccharomyces cerevisiae is the result of a missense mutation in the SUP35 gene. Genetics 1994; 137:659 - 670
- Young C, Cox B. Extrachromosomal elements in a super-suppression system of yeast I. A nuclear gene controlling the inheritance of the extrachromosomal elements. Heredity 1971; 26:413 - 422
- DiSalvo S, Derdowski A, Pezza JA, Serio TR. Dominant-negative prion mutants induce curing through distinct pathways that promote chaperone-mediated aggregate disassembly. Nature Struct Mol Biol 2011; 18:486 - 492
- Bossers A, Belt P, Raymond GJ, Caughey B, de Vries R, Smits MA. Scrapie susceptibility-linked polymorphisms modulate the in vitro conversion of sheep prion protein to protease-resistant forms. Proc Natl Acad Sci USA 1997; 94:4931 - 4936
- Kochneva-Pervukhova NV, Paushkin SV, Kushnirov VV, Cox BS, Tuite MF, Ter-Avanesyan MD. Mechanism of inhibition of [PSI+] prion determinant propagation by a mutation of the N-terminus of the yeast Sup35 protein. EMBO J 1998; 17:5805 - 5810
- Osherovich LZ, Cox BS, Tuite MF, Weissman JS. Dissection and design of yeast prions. PLoS Biol 2004; 2:86
- Luheshi LM, Dobson CM. Bridging the gap: from protein misfolding to protein misfolding diseases. FEBS Lett 2009; 583:2581 - 2586
- Bellotti V, Chiti F. Amyloidogenesis in its biological environment: challenging a fundamental issue in protein misfolding diseases. Curr Opin Struct Biol 2008; 18:771 - 779
- Verges KJ, Smith MH, Toyama BH, Weissman JS. Strain conformation, primary structure and the propagation of the yeast prion [PSI+]. Nat Struct Mol Biol 2011; 18:493 - 499