Abstract
The prion protein is a glycoprotein characterized by a folded -helical structure that, under pathological conditions, misfolds and aggregates into its infectious isoform as -sheet rich amyloidic deposits. The accumulation of the abnormal protein is responsible for a group of progressive and fatal disorders characterized by vacuolation, gliosis, and spongiform degeneration. Prion disorders are characterized by a triple aetiology: familial, sporadic or acquired, although most cases are sporadic. The mechanisms underlying prion neurotoxicity remain controversial, while novel findings lead to hypothesize intriguing pathways responsible for prion spreading. The present review aims to examine the involvement of the gastrointestinal tract and hypothesizes the potential mechanisms underlying cell-to-cell transmission of the prion protein. In particular, a special emphasis is posed on the mechanisms of prion transmission within the gut and towards the central nervous system. The glycation of prion protein to form advanced glycation end-products (AGE) interacting with specific receptors placed on neighboring cells (RAGE) represents the key hypothesis to be discussed.
Introduction
The physiological role of PrPc in healthy tissues still remains to be fully clarified. However, PrPc is involved in neuroprotection, signal transduction, suppression of apoptosis, neuronal development, long-term memory formation, haematopoietic stem cell self-renewal, cell adhesion, copper buffer and copper reductase activity.Citation1,Citation2
PrPc is characterized by a folded α-helical structure. Under pathological conditions, the prion protein misfolds and aggregates into its infectious isoform, PrPSc forming β-sheet rich amyloidic deposits. The accumulation of the abnormal protein characterizes transmissible spongiform encephalopathies (TSEs) or prion diseases, characterized by vacuolation, gliosis and spongiform degeneration. The human forms of this pathology include: the Creutzfeld-Jacob disease (CJD) with its sporadic, familial and new variant types; the rare and familial Gerstmann-Sträussler-Scheinker syndrome; the fatal familial insomnia; the epidemic kuru due to cannibalism practices.
According to the widely accepted “protein only” hypothesis proposed by Prusiner,Citation3 TSEs are caused by the conversion of the normal prion protein PrPc into its infectious conformational isoform PrPSc, which acting as a template, induces its own replication by an autocatalytic process and causes a further conformational change of other normal prion proteins.
This hypothesis, initially proposed to explain the infectious origin of the prion disorders, was re-modulated by Fornai et al.Citation4 These authors, focusing on the most recent findings suggested a common pathogenesis for the infectious, sporadic and familial forms of prion diseases. In particular, moving from the evidence that the “infectious” PrPSc is not toxic per se,Citation5 the unifying hypothesis suggests that neurotoxicity which characterizes prion disorders does not require PrPSc as the primary event. In contrast, the critical step may consist in the accumulation of prion protein. This may preceed PrPSc aggregation in a variety of conditions. In fact, possible reasons for accumulation of PrPSc-enriched proteinaceous aggregates is the presence of a genetically abnormal protein, a normal protein that changes its structure because of an “infectious” template, or an absolute/relative excess of normal protein.Citation6 In addition, it might be hypothesized that exogenous factors such as bacterial or viral pathogens may impair the physiological clearance and/or intracellular trafficking of the protein through proteasome and mainly autophagy, thus triggering the aggregation of misfolded pathogenic prion protein.Citation4,Citation7
This latter model of PrPSc formation may explain some prion disorders. In line with this view, Sandberg et al.Citation8 recently proposed a new model of prion pathology based on uncoupling of infectivity and toxicity, which distinguishes two phases: (1) a clinically silent exponential phase which rapidly reaches a maximal prion titre and (2) a plateau phase which leads to the clinical onset depending on the amount of prion protein concentration. This model hypothesizes that during the template-assisted progression from PrPc to PrPSc an intermediate toxic species, named PrPl (lethal PrP), is formed when prion propagation saturates, leading to a switch from autocatalytic production of infectivity (phase 1) to a toxic (phase 2) pathway.
However, a question still remains unsolved: which is the molecular mechanism underlying cell-to-cell transmission of PrPSc? In other words, how does the propagation of PrPSc occur from pathological “infected” to normal tissues?
In experimental studies, inoculation of the infected brain homogenates directly into the brain of healthy animals is commonly used. The rationale is that oral exposure to the infected material is relatively inefficient and commonly results in longer incubation periods.
More recently, Makarawa et al.Citation9 demonstrated that transmissible prion diseases can be induced in wild-type animals by inoculation of recombinant prion protein. This finding is relevant in order to disclose the mechanisms and the pathways involved in the PrPSc transmission. In fact, since in nature prion diseases are usually transmitted by extracerebral prion infection, mainly involving the oral transmission, the knowledge of the exact sequence of events which starts with ingestion of the infectious material and leads to central nervous system (CNS) invasion is relevant.Citation10
In particular, oral transmission raises a number of questions related to the sequence of events which starts with ingestion of the infectious material.
The present review aims to hypothesize the dynamics of cell-to-cell transmission of the prion protein in the gastrointestinal (GI) tract and toward the CNS. These hypotheses are based on facts and analogies and call for specific experiments aimed at establishing the reliability of such a hypothetical model in order to design possible therapeutic strategies which interrupt successfully the journey of prion proteins to CNS.
Entering the Gastrointestinal Tract
The prion protein is normally expressed, mainly in neurons and lymphoid tissues, but the physiological role is not completely understood. It seems that alterations and/or accumulation of prion are responsible for the onset of TSEs. However, the mechanisms responsible for neurotoxicity remain under debate and beyond the scope of the present review, which focuses on potential prion transmission pathways. Since in nature prion diseases are usually transmitted by extracerebral prion infection, understanding the mechanisms and the pathways leading to CNS invasion is relevant. It is well-established that a conditio sine qua non for spreading the prion infection to the CNS is the presence of the prion protein in peripheral tissues, and in particular the GI tract.Citation5 In this respect, PrPc was found to be constitutively expressed in both human and animal enteric nervous system (ENS), as well as in epithelial and glandular cells throughout the GI tract.Citation10,Citation11 A recent study showed that in mice the glycosylphosphatidylinositol-anchored membrane PrPc form is essential for the rapid neural spread and CNS invasion of prion infection, with respect to the anchorless PrPc.Citation12 Some mechanisms of neuroinvasion are well-documented, while others remain putative.
It is established that the protease-resistance of PrPSc because of its β-sheet structure allows the PrPSc to easily enter the intestinal epithelium and reach the ENS in the oral transmission route. At the epithelial level, three cell types are considered the site of entry: villous columnar epithelial cells (enterocytes), dendritic cells (DCs) and microfold (M) cells. Intestinal epithelial cells can transcytose fragments of PrPSc complexed with ferritin: once translocated at the basolateral side of the enterocytes, PrPSc can be accumulated and degraded by macrophages or transported to lymphoid tissues by DCs.Citation13,Citation14 Ano et al.Citation15 showed that PrPSc was found to be incorporated into the enterocytes and was also detected in the villous lacteal in suckling, but not weaned, mice, suggesting that maternal immunoglobulins are important risk factors for the oral transmission of PrPSc. Interestingly, in humans the protease resistant PrPSc is transcytosed across enterocytes independently of endogenous PrPc expression, suggesting an alternative receptor-mediated prion uptake mechanism. In this respect, the 37-kDa/67-kDa laminin receptor (LRP/LR), expressed on the apical brush border of enterocytes, is responsible for the binding of both PrPc and infectious prions. It is likely that LRP/LR might act as a key player in the intestine allowing prions of some species to pass and prions of other species not to pass. In fact, Morel et al.Citation16 showed that bovine spongiform encephalopathy (BSE) prions become internalized by human enterocytes dependent on LRP/LR and might have caused the zoonotic variant CJD. Kolodiejczak et al.Citation17 extended the study mainly employing co-localization studies between prions of different species and LRP/LR on different enterocyte species, suggesting that interspecies prion infection is strongly dependent on the specific interactions between prions and LRP/LR.Citation17 The main suggestion by these authors was that chronic wasting disease (CWD) prions and scrapie prions co-localize with LRP/LR on human enterocytes and might therefore cause other human prion diseases. An exhaustive revision of the proposed role of the 37 kDa/67 kDa laminin receptor in the intestinal pathophysiology of alimentary prion infections as “natural” mechanism of prion transmission, was recently carried out by Da Costa Dias et al.Citation18
The migratory bone marrow-derived DCs are usually localized beneath M cells from which they receive PrPSc to be delivered to lymphoid tissue, but they can also catch PrPSc directly from the intestinal lumen by opening the tight junctions of enterocytes and by inserting their dendrites between them. Unlike macrophages, some DCs can retain PrPSc in its infectious non-degraded state.Citation13
One of the main candidates for the entry of PrPSc across the intestinal epithelium is supposed to be the M cell which is specialized for the transepithelial transport of macromolecules to enable the immune system to sample the content of the gut lumen. Thanks to this property, M cells were shown to transport PrPSc through the basolateral side of the epithelium, where they possess a special pocket.Citation19 The role of M cells has been recently confirmed in an in vitro bovine M cell model by co-culture with murine intestinal lymphocytes. In this model, M cell-differentiated bovine intestinal epithelial cells were able to deliver the infectious agents at least 30-fold more efficiently than undifferentiated cells.Citation20
The next step for PrPSc transmission before neuroinvasion is the gut-associated lymphoid tissue (GALT), including Peyer's patches and mesenteric lymph nodes (but also, spleen, tonsils), where PrPSc accumulates and it is amplified. Interestingly, high levels of PrPSc are expressed within germinal centres of B-cell follicles, in particular on fixed, stromal-derived follicular dendritic cells (FDCs). As a matter of fact, lymphoid tissue appears essential for the onset of prion disease, since an absence of Peyer's patches, and especially of FDCs, impairs or delays neuroinvasion after oral inoculation of PrPSc.Citation13,Citation14
Three kinds of cells can contact the pocket of M cells and then can be possibly responsible for PrPSc transport and progression: lymphocytes, macrophages and DCs. Lymphocytes are unlike to be involved, since they do not seem to acquire significant levels of PrPSc following intraintestinal exposure. The role of macrophages is still uncertain, but these cells seem to impair early accumulation of PrPSc.Citation13,Citation14 In fact, in a mouse model of infection, gut macrophages have been implicated in the initial clearance of infectious particles as well as in later steps of the propagation process.Citation21 Unlike lymphocytes and macrophages, DCs appear responsible for delivering PrPSc to GALT. These cells are also able to acquire PrPSc independently of M cells by interrupting epithelial tight junction, as described before. Once inside the DC, the protein is processed into lysosomes and short peptides are finally presented on the cell surface in association with the major histocompatibility complex class II molecules. Then, some DCs, like macrophages, cleave PrPSc by cysteine proteases, but other DCs can retain prion protein in its infectious form and deliver it to GALT.Citation22 However, the mechanism through which DCs propagate PrPSc to FDCs remains unclear. In fact, DCs migrate to the T-cell region of GALT which is anatomically distinct from the B-cell follicles where FDCs reside. Neverthless, a subset of DCs retaining the infectious particles has been found to interact with B cells. Furthermore, the complement system might also favour PrPSc uptake.Citation13,Citation14 After invasion, accumulation and amplification in the GALT, PrPSc spreads to ENS.
From Enteric Neuroinvasion to Central Nervous System
Experimental data in rodents suggest that after oral inoculation, PrPSc spreads from GALT to the CNS via the ENS.Citation23 The infectious agent moves to ENS when axons or dendrites interact with GALT or epithelial cells. Two routes involving the two sections of the autonomic nervous system have been hypothesized to explain the retrograde spreading of prions from the GI tract to the CNS: (1) the sympathetic coeliac and mesenteric ganglion complex, through splanchnic nerves and (2) the lumbal spinal cord or parasympathetic vagus nerve. In line with this, neuroinvasion does not occur if sympathetic nerves are impaired.Citation24 On the other hand, the dorsal root ganglia are not essential, and prion invasion of the non-autonomic peripheral nervous system appears a secondary event which might follow prion replication within CNS.Citation25
Finally, it must be taken into account that prion infection of the ENS might occur within fine nerve fibers directly underneath the villous or glandular epithelium.Citation26 Moreover, prions are able to spread from the small intestine to the oral salivary glands and epithelia, a route which might explain the occurrence of prions in the saliva and the shedding of prions into the oral cavity at least of sheep.Citation27,Citation28
Neuroinvasion correlates with the placement of prions within FDCs and sympathetic nerves and would occur rapidly from sites where FDCs are closely associated with nerve fibers, such as GALT.Citation13,Citation14 In fact, FDCs might be able to transfer directly PrPSc to ENS, since anatomical connections between these cells and nervous terminals have been described under electron microscopy.Citation29 DCs also establish contacts with neurites of peripheral neurons through cytoplasmic extensions.Citation30
The innervation of the Peyer's patches in the sheep ileum suggests that PrPSc may colonize both intrinsic and extrinsic neurons, the subsequent neuroinvasion towards CNS being due to the intrinsic Dogiel type II neurons.Citation31 Not surprisingly, immunolabeling for PrPc in mice was found to be more pronounced in enteric glial cells than enteric neurons, suggesting glia as a potential site of replication for infectious prions.Citation11
On the other hand, the transmission of PrPSc from CNS towards the periphery is also described in reference Citation32. In a recent paper, PrPSc damaging enteric glial cells and Peyer's patches was found following intracerebral inoculation of prions. This also led to selective neuronal loss in the enteric wall (including nitric oxide synthase- and tyrosine hydroxylase-positive neurons).Citation33 The authors hypothesize that prions travel from the periphery to CNS and viceversa through neuronal pathways, as well as blood stream.
This scenario calls to hypothesize that centripetal and centrifugal propagation of PrPSc needs a cell-to-cell spreading, similarly to what was recently demonstrated for a variety of misfolded prion-like proteins,Citation34 which are secreted from cells into the extracellular environment in the form of exosomes, and might be taken up by receptors for advanced glycation end-products (RAGE), in the neighboring cells. In keeping with this, the prion protein released in the extracellular space can initiate infection in both neuronal and non-neuronal cells.Citation35,Citation36
Molecular Mechanisms of Cell-to-Cell Spreading of Prion Protein
In order to understand the prion spreading, it is important to know how membrane-anchored PrPc is internalized in the cell. At present, different mechanisms have been proposed. First, LRP/LR has been shown to act as a receptor for cellular PrP on the surface of mammalian cellsCitation37 and is responsible for PrPc internalization. In fact, PrPc can be internalized via the classic pathway involving clathrin-coated pits-mediated endocytosis. Moreover, membrane lipid rafts, in particular caveolae-like domains, have also been implicated, and they appear important for PrPc trafficking, since their disruption by sterol-binding agents, such as filipin or nystatin, inhibits cupper-stimulated PrPc internalization. The recent observation that clathrin depletion does non affect PrPc internalization led to the discovery that a clathrin-independent mechanism can involve the endocytic pathway associated with Arf6.Citation38 A similar clathrin-independent internalization is shared by virusCitation39 and other proteins belonging to the family of glycosylphosphatidylinositol-anchored proteins.Citation40
Therefore, the presence of the glycosylphosphatidylinositol domain confers to the physiological form of PrPc specific properties which allow the protein to be transmitted, as first shown by Baron et al.Citation41 It is likely that such an anchor allows the protein to behave like an infectious agent (i.e., a viral particle or an intracellular bacterium), which needs to be internalized by the cell to replicate and propagate towards other cells.Citation4
Increasing evidence indicates that the intercellular transfer of membrane patches, containing membrane-anchored proteins from one cell type to another, named trogocytosis and classically described for immune cells, is a more general mechanism of cell-to-cell communication, which in particular involves surface membranes containing glycosylphosphatidylinositol-anchored proteins.Citation42 In this view, we might hypothesize that the mechanism of cell-to-cell propagation of prions involves trogocytosis, which drives the spreading of the prion within the gut and towards the CNS.
Another important feature to be addressed is the role of the clearing pathways in the PrPSc transferring from GALT cells to peripheral nerves of the ENS. Similarly to what happens to proteinaceous aggregates in other NDs, such as synucleinopathies, Alzheimer disease, amyotrophic lateral sclerosis and Huntington disease, PrPSc shares the property to be metabolized by the autophagy pathway. However, the role of autophagy in prion diseases is still controversial. In fact, although there is good evidence that induction of autophagy is beneficial in prion-infected cells and animals, it is not clear whether increased or decreased autophagy have deleterious effects. From one hand, there is direct evidence that induction of autophagy results in degradation of cellular PrPSc, and cells impaired in autophagy might be more susceptible to prion infection as they are lacking a putative defence mechanism. In fact, autophagy inducers such as lithium, trehalose, imatinib or rapamycin enhance the clearance of PrPSc in prion-infected cells.Citation43
On the other hand, autophagy might be a positive factor for prion propagation, since moderate autophagy might be beneficial for generating smaller PrPSc seeds. In fact, these seeds are considered more effective templates to convert PrPc into PrPSc with respect to larger aggregates. Therefore, a moderate level of autophagy might be essential to support PrPSc production during certain stages of prion infection and, consequently, to promote prion transmission.Citation44 Concerning the ubiquitin-proteasome system (UPS), it is not yet clear whether the impairment of this system is a primary or secondary event in prion disease pathophysiology.Citation45,Citation46 PrPSc can be processed by UPS only when it is soluble. Experimental evidence shows that altered PrPc metabolism, as a consequence of either a primary UPS inhibition or an excess of UPS substrates, produces increased amounts of cytosolic PrPc, followed by marked neurotoxicity.Citation47
The molecular steps by which autophagy clears damaged proteins and organelles within the cell are shown in . The interactions within autophagy and UPS and formation of a new membrane vacuole,Citation48,Citation49 named autophagoproteasomeCitation50–Citation52 are also shown.
Misfolded prion protein accumulates based on the efficacy of protein clearing mechanisms such as the autophagy pathway and the UPS. Therefore, an impairment of the autophagy system in ENS neurons may lead to the lack of clearance of such autophagy-dependent transmissible misfolded proteins, which, upon cell-to-cell spreading and progressive neuronal death, ultimately may reach trans-synaptically the CNS.
When not appropriately degraded by autophagy pathway or UPS, several misfolded proteins, such as β-amyloid (Aβ), copper zinc superoxide dismutase 1 (SOD-1), α-synuclein, TDP-43 and tau protein, can undergo spontaneous glycation as a post-translational, non-enzymatic reaction between reducing sugars and amino groups, named the Maillard reaction. These early glycated products undergo further complex and advanced glycation and oxidation (glycoxidation), which elicits irreversibile modification, to form heteromorphic and fluorescent derivatives with the formation of advanced glycation end-products (AGE). This process is responsible for an increased protein stability by the formation of crosslinks that stabilize protein aggregates.Citation2 Since the glycation of the prion protein has been shown to occur as a spontaneous process,Citation53 we pose the hypothesis that conversion of the prion protein to AGE also occurs, thus predisposing such a modified prion protein to be internalized by RAGE through a nystatin- and filipin-sensitive, clathrin-independent endocytosis (). This would contribute to the accumulation of aberrant proteins in the CNS. In line with these hypotheses, high amounts of glycated products co-localize with prion-positive plaques in the brain of CJD patients.Citation54
Glycation leading to AGE can be divided into two reaction stages:
(1) The early stage reaction starts with the interaction of reducing sugars, such as glucose. The reaction is reversible up to the point of the synthesis of the Amadori product and is dependent on compound concentrations and incubation time. (2) The late stage reaction is an irreversible reaction, involving dehydration, hydrolysis and consequently results in the formation of AGE ().
Immunostaining studies indicate that, at least in affected hamsters, astrocytes are the first site of this glycation process. Since glycation is accompanied by oxidation, oxidative stress also occurs, with the production of reactive oxygen species which may lead to cell death.Citation55,Citation56 AGE are expressed on the plasma membrane and may be targeted by specific antibodies. Once AGE are produced, they diffuse extracellularly to interact with neighbouring cells. The main pathway which is responsible for their exocytosis involves the formation of a vesicle, named exosome, which extrudes the glycated proteins in the extracellular space.
Two kinds of receptors are able to remove AGE: RAGE and scavenger receptors (SRs). In addition, three proteins have been reported as AGE-binding proteins: AGE-R1 (oligosaccharyl transferase complex protein 48: OST48), AGE-R2 (80K-H protein) and AGE-R3 (galectin-3). RAGE are a member of the immunoglobulin superfamily of cell-surface molecules, and they are expressed in a variety of cell types, including endothelial cells, smooth muscle cells, lymphocytes, monocytes, macrophages, DCs and neurons. SRs are homotrimeric cell-surface molecules and are expressed only on resident macrophages or microglial cells. RAGE and SR mediate cell adhesion to Aβ and induction of oxidative stress.Citation55
RAGE can exist in two prevalent isoforms: the full-length and the secreted RAGE isoform. The full-length isoform is composed of three extracellular immunoglobulin-like domains (V, C1 and C2), a single transmembrane spanning helix, and a short intracellular domain. The secreted isoform, as its name indicates, lacks the transmembrane and the intracellular domains and it is so free in the extracellular space where it is thought to play the role of a decoy that interacts with free circulating RAGE ligand.Citation57
The binding of AGE to their receptor activates multiple signaling pathways: RAGE-mediated activation of protein kinase C upregulates Egr-1 expression, while stimulation of ras and src kinase is a key step in activation of NFκB. Moreover, NFκB activation leads to a further increase in RAGE expression by a positive feedback mechanism. Such a vicious circle, joined with upregulation of Egr-1 and NFκB, induces oxidative stress and inflammation causing cell damage up to neuronal death in the enteric wall. These mechanisms operate in addition to cell-to-cell transmission and cause both protein spreading and trans-synaptic neuronal loss.Citation57,Citation58
AGE can be eliminated by specific antibodies or can be propagated via RAGE interaction.Citation2 Recently, internalization and self-sustaining spreading of AGE was described in the ENS, where AGE produce the loss of GI neurons.Citation59 In this way, the propagation of the degenerative process may enter the nerves to reach the CNS.
In CJD patients, many prion-positive plaques immunoreactive for the anti-AGE antibody were observed, and many astrocytes were found to contain prion-, AGE- and RAGE-immunopositive granules, thus suggesting that there may be a RAGE-mediated PrP degradation pathway in CJD similar to what observed also in the case of Aβ protein in Alzheimer disease.Citation54
This intriguing hypothesis calls for specific experiment aimed at exploring this mechanism.
Abbreviations
| Aβ | = | β-amyloid |
| AGE | = | advanced glycation end-products |
| CNS | = | central nervous system |
| DCs | = | dendritic cells |
| ENS | = | enteric nervous system |
| FDCs | = | follicular dendritic cells |
| GALT | = | gut-associated lymphoid tissue |
| GI | = | gastrointestinal |
| LRP/LR | = | 37 kDa/67 kDa laminin receptor |
| M cells | = | microfold cells |
| MP | = | myenteric plexus |
| NDs | = | neurodegenerative diseases |
| PrPc | = | normal prion protein |
| PrPSc | = | altered prion protein |
| RAGE | = | receptors for advanced glycation end-products |
| SMP | = | submucosal plexus |
| SOD-1 | = | copper zinc superoxide dismutase 1 |
| SRs | = | scavenger receptors |
| TSEs | = | transmissible spongiform encephalopathies |
| UPS | = | ubiquitin-proteasome system |
Figures and Tables
Figure 1 The autophagy pathway may converge with the proteasome to produce a powerful protein clearing system named autophagoproteasome. Most of the protein aggregates which occur in the gut mirror analogous structures within CNS. These proteins all share the property to be metabolized preferentially by the autophagy pathway. This powerful protein and organelles clearing system starts mainly in the membrane of the Golgi apparatus and endoplasmic reticulum where it blossoms as a primary structure named the phagophore which becomes committed to the autophagy pathway following the association with the protein LC3-II. This multi-membrane vesicle develops as an autophagosome which is characterized by a double membrane vesicle in which mono or poly ubiquitinated misfolded proteins and/or mitochondria are sequestered. The autophagosome may directly fuse with the lysosome or merge with the late endosome (known also as multi vesicular body) to build up an amphisome which eventually merges with the lysosome. Another protein clearing pathway is the ubiquitin proteasome system which clears only protein substrate upon their polyubiquitination. Despite being described as distinct organelles in the cell, recent experimental evidence indicates that proteasome components and autophagosome converge into a complex clearing organelle in which the proteasome enzymes add on classic autophagy proteins,Citation48,Citation49 which was named the autophagoproteasome.Citation50–Citation52
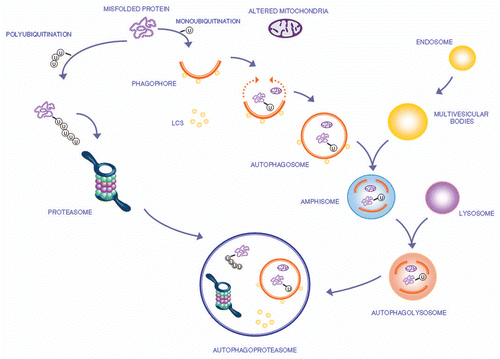
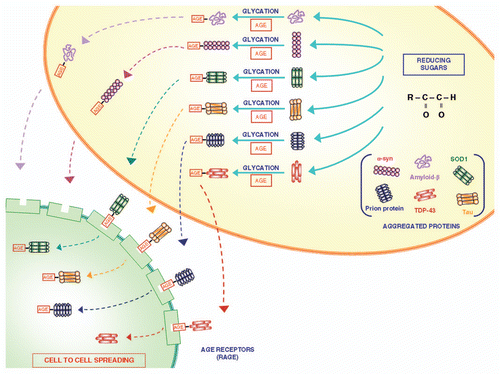
Figure 2 The role of glycation in modifying proteins structure and function. Glycation is a post-translational, non-enzymatic reaction between reducing sugars and amino groups, resulting in the formation of AGE. The major glycation agents include methylglyoxal, glyoxal and 3-deoxyglucosone. Misfolded proteins represent an important target for glycation. A variety of proteins involved in neurodegenerative disorders (such as Aβ, SOD-1, α-synuclein, TDP-43, tau protein and prion protein) undergo glycation, responsible for an increased stability by the formation of crosslinks that stabilize protein aggregates. These AGE are expressed on the plasma membrane and may diffuse extracellularly to interact with neighbouring cells to spread according to a prion-like mechanism.Citation34 In fact, glycated proteins would interact with RAGE, thus allowing a “cell to cell spreading” by their internalization. This process, which is well demonstrated within CNS, remains at hypothetical level within the gut, where in any case the prion protein is shown to spread from cell to cell.
References
- Martins VR, Mercadante AF, Cabral AL, Freitas AR, Castro RM. Insights into the physiological function of cellular prion protein. Braz J Med Biol Res 2001; 34:585 - 595
- Vicente Miranda H, Outeiro TF. The sour side of neurodegenerative disorders: the effects of protein glycation. J Pathol 2010; 221:13 - 25
- Prusiner SB. Novel proteinaceous infectious particles cause scrapie. Science 1982; 216:136 - 144
- Fornai F, Ferrucci M, Gesi M, Bandettini di Poggio A, Giorgi FS, Biagioni F, et al. A hypothesis on prion disorders: are infectious, inherited and sporadic causes so distinct?. Brain Res Bull 2006; 69:95 - 100
- Davies GA, Bryant AR, Reynolds JD, Jirik FR, Sharkey KA. Prion diseases and the gastrointestinal tract. Can J Gastroenterol 2006; 20:18 - 24
- Nicolas O, Gavín R, del Río JA. New insights into cellular prion protein (PrPc) functions: the “ying and yang” of a relevant protein. Brain Res Rev 2009; 61:170 - 184
- Aguzzi A, Hardt WD. Dangerous liaisons between a microbe and the prion protein. J Exp Med 2003; 198:1 - 4
- Sandberg MK, Al-Doujaily H, Sharps B, Clarke AR, Collinge J. Prion propagation and toxicity in vivo occur in two distinct mechanistic phases. Nature 2011; 470:540 - 542
- Makarava N, Kovacs GG, Bocharova O, Savtchenko R, Alexeeva I, Budka H, et al. Recombinant prion protein induces a new transmissible prion disease in wild-type animals. Acta Neuropathol 2010; 119:177 - 187
- Shmakov AN, Ghosh S. Prion proteins and the gut: une liaison dangereuse?. Gut 2001; 48:443 - 447
- Albanese V, Lawson VA, Hill AF, Cappai R, Di Guardo G, Staikopoulos V, et al. Evidence for prion protein expression in enteroglial cells of the myenteric plexus of mouse intestine. Auton Neurosci 2008; 140:17 - 23
- Klingeborn M, Race B, Meade-White KD, Rosenke R, Striebel JF, Chesebro B. Crucial role for prion protein membrane anchoring in the neuroinvasion and neural spread of prion infection. J Virol 2011; 85:1484 - 1494
- Mabbott NA, MacPherson GG. Prions and their lethal journey to the brain. Nat Rev Microbiol 2006; 4:201 - 211
- Ano Y, Sakudo A, Nakayama H, Onodera T. Uptake and dynamics of infectious prion protein in the intestine. Protein Pept Lett 2009; 16:247 - 255
- Ano Y, Sakudo A, Uraki R, Sato Y, Kono J, Sugiura K, et al. Enhanced enteric invasion of scrapie agents into the villous columnar epithelium via maternal immunoglobulin. Int J Mol Med 2010; 26:845 - 851
- Morel E, Andrieu T, Casagrande F, Gauczynski S, Weiss S, Grassi J, et al. Bovine prion is endocytosed by human enterocytes via the 37 kDa/67 kDa laminin receptor. Am J Pathol 2005; 167:1033 - 1042
- Kolodziejczak D, Da Costa Dias B, Zuber C, Jovanovic K, Omar A, Beck J, et al. Prion interaction with the 37-kDa/67-kDa laminin receptor on enterocytes as a cellular model for intestinal uptake of prions. J Mol Biol 2010; 402:293 - 300
- Da Costa Dias B, Jovanovic K, Weiss SF. Alimentary prion infections: Touchdown in the intestine. Prion 2011; 5:6 - 9
- Heppner FL, Christ AD, Klein MA, Prinz M, Fried M, Kraehenbuhl JP, et al. Transepithelial prion transport by M cells. Nat Med 2001; 7:976 - 977
- Miyazawa K, Kanaya T, Takakura I, Tanaka S, Hondo T, Watanabe H, et al. Transcytosis of murine-adapted bovine spongiform encephalopathy agents in an in vitro bovine M cell model. J Virol 2010; 84:12285 - 12291
- Maignien T, Shakweh M, Calvo P, Marcé D, Salès N, Fattal E, et al. Role of gut macrophages in mice orally contaminated with scrapie or BSE. Int J Pharm 2005; 298:293 - 304
- Huang FP, Farquhar CF, Mabbott NA, Bruce ME, MacPherson GG. Migrating intestinal dendritic cells transport PrP(Sc) from the gut. J Gen Virol 2002; 83:267 - 271
- McBride PA, Schulz-Schaeffer WJ, Donaldson M, Bruce M, Diringer H, Kretzschmar HA, et al. Early spread of scrapie from the gastrointestinal tract to the central nervous system involves autonomic fibers of the splanchnic and vagus nerves. J Virol 2001; 75:9320 - 9327
- Glatzel M, Heppner FL, Albers KM, Aguzzi A. Sympathetic innervation of lymphoreticular organs is rate limiting for prion neuroinvasion. Neuron 2001; 31:25 - 34
- Hoffmann C, Ziegler U, Buschmann A, Weber A, Kupfer L, Oelschlegel A, et al. Prions spread via the autonomic nervous system from the gut to the central nervous system in cattle incubating bovine spongiform encephalopathy. J Gen Virol 2007; 88:1048 - 1055
- Jeffrey M, González L, Espenes A, Press CM, Martin S, Chaplin M, et al. Transportation of prion protein across the intestinal mucosa of scrapie-susceptible and scrapie-resistant sheep. J Pathol 2006; 209:4 - 14
- Maddison BC, Rees HC, Baker CA, Taema M, Bellworthy SJ, Thorne L, et al. Prions are secreted into the oral cavity in sheep with preclinical scrapie. J Infect Dis 2010; 201:1672 - 1676
- Da Costa Dias B, Weiss SF. A kiss of a prion: new implications for oral transmissibility. J Infect Dis 2010; 201:1615 - 1616
- Heggebø R, Gonzàlez L, Press CM, Gunnes G, Espenes A, Jeffrey M. Disease-associated PrP in the enteric nervous system of scrapie-affected Suffolk sheep. J Gen Virol 2003; 84:1327 - 1338
- Dorban G, Defaweux V, Heinen E, Antoine N. Spreading of prions from the immune to the peripheral nervous system: a potential implication of dendritic cells. Histochem Cell Biol 2010; 133:493 - 504
- Chiocchetti R, Mazzuoli G, Albanese V, Mazzoni M, Clavenzani P, Lalatta-Costerbosa G, et al. Anatomical evidence for ileal Peyer's patches innervation by enteric nervous system: a potential route for prion neuroinvasion?. Cell Tissue Res 2008; 332:185 - 194
- Kimberlin RH, Field HJ, Walker CA. Pathogenesis of mouse scrapie: evidence for spread of infection from central to peripheral nervous system. J Gen Virol 1983; 64:713 - 716
- Lawson VA, Furness JB, Klemm HM, Pontell L, Chan E, Hill AF, et al. The brain to gut pathway: a possible route of prion transmission. Gut 2010; 59:1643 - 1651
- Lee SJ, Desplats P, Sigurdson C, Tsigelny I, Masliah E. Cell-to-cell transmission of non-prion protein aggregates. Nat Rev Neurol 2010; 6:702 - 706
- Wang G, Zhou X, Bai Y, Zhang Z, Zhao D. Cellular prion protein released on exosomes from macrophages binds to Hsp70. Acta Biochim Biophys Sin (Shanghai) 2010; 42:345 - 350
- Gousset K, Schiff E, Langevin C, Marijanovic Z, Caputo A, Browman DT, et al. Prions hijack tunnelling nanotubes for intercellular spread. Nat Cell Biol 2009; 11:328 - 336
- Gauczynski S, Peyrin JM, Haïk S, Leucht C, Hundt C, Rieger R, et al. The 37-kDa/67-kDa laminin receptor acts as the cell-surface receptor for the cellular prion protein. EMBO J 2001; 20:5863 - 5875
- Kang YS, Zhao X, Lovaas J, Eisenberg E, Greene LE. Clathrin-independent internalization of normal cellular prion protein in neuroblastoma cells is associated with the Arf6 pathway. J Cell Sci 2009; 122:4062 - 4069
- Heikkilä O, Susi P, Tevaluoto T, Härmä H, Marjomäki V, Hyypiä T, et al. Internalization of coxsackievirus A9 is mediated by {beta}2-microglobulin, dynamin and Arf6 but not by caveolin-1 or clathrin. J Virol 2010; 84:3666 - 3681
- Donaldson JG, Porat-Shliom N, Cohen LA. Clathrin-independent endocytosis: A unique platform for cell signaling and PM remodeling. Cell Sig 2009; 21:1 - 6
- Baron GS, Wehrly K, Dorward DW, Chesebro B, Caughey B. Conversion of raft associated prion protein to the protease-resistant state requires insertion of PrP-res (PrP(Sc)) into contiguous membranes. EMBO J 2002; 21:1031 - 1040
- Ahmed KA, Munegowda MA, Xie Y, Xiang J. Intercellular trogocytosis plays an important role in modulation of immune responses. Cell Mol Immunol 2008; 5:261 - 269
- Heiseke A, Aguib Y, Riemer C, Baier M, Schätzl HM. Lithium induces clearance of protease resistant prion protein in prion-infected cells by induction of autophagy. J Neurochem 2009; 109:25 - 34
- Heiseke A, Aguib Y, Schatzl HM. Autophagy, prion infection and their mutual interactions. Curr Issues Mol Biol 2010; 12:87 - 97
- Deriziotis P, Tabrizi SJ. Prions and the proteasome. Biochim Biophys Acta 2008; 1782:713 - 722
- Whatley BR, Li L, Chin LS. The ubiquitin-proteasome system in spongiform degenerative disorders. Biochim Biophys Acta 2008; 1782:700 - 712
- Ma J, Wollmann R, Lindquist S. Neurotoxicity and neurodegeneration when PrP accumulates in the cytosol. Science 2002; 298:1781 - 1785
- Castino R, Lazzeri G, Lenzi P, Bellio N, Follo C, Ferrucci M, et al. Suppression of autophagy precipitates neuronal cell death following low doses of methamphetamine. J Neurochem 2008; 106:1426 - 1439
- Lenzi P, Fulceri F, Lazzeri G, Casini A, Ruggieri S, Paparelli A, et al. Analysis of single, purified inclusions as a novel approach to understand methamphetamine neurotoxicity. Ann NY Acad Sci 2008; 1139:186 - 190
- Pasquali L, Ruggieri S, Murri L, Paparelli A, Fornai F. Does autophagy worsen or improve the survival of dopaminergic neurons?. Parkinsonism Relat Disord 2009; 15:24 - 27
- Isidoro C, Biagioni F, Giorgi FS, Fulceri F, Paparelli A, Fornai F. The role of autophagy on the survival of dopamine neurons. Curr Top Med Chem 2009; 9:869 - 879
- Ferrucci M, Fulceri F, Toti L, Soldani P, Siciliano G, Paparelli A, et al. Protein clearing pathways in ALS. Arch Ital Biol 2011; 149:121 - 149
- Panza G, Dumpitak C, Birkmann E. Influence of the maillard reaction to prion protein aggregation. Rejuvenation Res 2010; 13:220 - 223
- Sasaki N, Takeuchi M, Chowei H, Kikuchi S, Hayashi Y, Nakano N, et al. Advanced glycation end products (AGE) and their receptor (RAGE) in the brain of patients with Creutzfeldt-Jakob disease with prion plaques. Neurosci Lett 2002; 326:117 - 120
- Kikuchi S, Shinpo K, Takeuchi M, Yamagishi S, Makita Z, Sasaki N, et al. Glycation—a sweet tempter for neuronal death. Brain Res Brain Res Rev 2003; 41:306 - 323
- Choi YG, Kim JI, Jeon YC, Park SJ, Choi EK, Rubenstein R, et al. Nonenzymatic glycation at the N terminus of pathogenic prion protein in transmissible spongiform encephalopathies. J Biol Chem 2004; 279:30402 - 30409
- Leclerc E, Sturchler E, Vetter SW. The S100B/RAGE axis in Alzheimer's disease. Cardiovasc Psychiatry Neurol 2010; 2010:539 - 581
- Xu Y, Toure F, Qu W, Lin L, Song F, Shen X, et al. Advanced glycation end product (AGE)-receptor for AGE (RAGE) signaling and upregulation of Egr-1 in hypoxic macrophages. J Biol Chem 2010; 285:23233 - 23240
- Jeyabal PV, Kumar R, Gangula PR, Micci MA, Pasricha PJ. Inhibitors of advanced glycation end-products prevent loss of enteric neuronal nitric oxide synthase in diabetic rats. Neurogastroenterol Motil 2008; 20:253 - 261