Abstract
Senile plaques composed of amyloid-beta (Aβ) in the brain are one of the hallmarks of Alzheimer disease (AD). Removal of Aβ from the brain is the most important therapeutic strategy for AD. The solubility of Aβ is critical for its endocytosis, transcytosis and removal from the brain. Our recent study has found that the extracellular domain of p75NTR, the neurotrophin receptor, plays an important role in the solubility of Aβ and might be one of the endogenous mechanisms in the regulation of Aβ plaque formation. The physiologically shedded extracellular domain of p75NTR is able to inhibit Aβ aggregation and diasggregate preformed Aβ fibrils, while the full p75NTR expressed on neurites, endothelial cells and smooth muscle cells in blood-brain barrier (BBB) might initiate Aβ endocytosis and degradation, and/or remove Aβ from the brain via BBB. Understanding the roles of p75NTR in the solubility and clearance of Aβ may allow targetting p75NTR as a unique opportunity to develop therapeutic drugs for the prevention and treatment of AD.
Senile plaque in the brain is one of the hallmarks of Alzheimer disease (AD). The main component of the senile plaques is amyloid-beta (Aβ), which is a metabolic product of amyloid precursor protein (APP). The steady-state level of Aβ in the normal brain is maintained by the balance between its production and clearance. However in the AD brain this balance is broken due to either over-production of Aβ or a reduction in Aβ clearance;Citation1,Citation2 thus Aβ accumulates in the brain and forms amyloid plaques which cause dementia and neurodegeneration in patients. Based on the Aβ hypothesis proposed a decade ago, Aβ plays a causal and pivotal role in the development of AD.Citation3 Therefore, removal of Aβ from the brain is the most important therapeutic strategy for AD.Citation4 To reach this goal, it is essential to understand how the Aβ metabolism is regulated in the AD brain. Despite the dramatic progress has been made in the understanding of how Aβ is produced from APP, the mechanisms of Aβ aggregation, metabolism and clearance from the brain remain unclear so far. Only 5% of AD (familial cases) is due to the over-production of Aβ because of mutations in the APP gene or in the APP processing enzymes, while the majority (95%) of so-called sporadic or late-onset AD (LOAD) are likely caused by dysfunctions in Aβ solubility or aggregation, endocytosis, degradation, transcytosis and removal.
The solubility of Aβ is critical for its endocytosis, transcytosis and removal from the brain. In AD patients, one of the most consistent biomarkers found so far is the reduction of Aβ level in the cerebral spinal fluid (CSF).Citation5 This is the most convincing evidence that there is a reduction in the solubility of Aβ and an increase in the Aβ aggregation and beta-sheet formation in AD patients. In sporadic cases of AD, the polymorphism of the Aβ-binding protein ApoE4 is highly associated with AD. It is known that other variants of ApoE proteins have a higher binding affinity to Aβ than ApoE4. It is likely that ApoE protein plays a critical role in the solubility of Aβ.Citation6 The reduction in the Aβ binding ability of ApoE4 may reduce the solubility of Aβ. Thus ApoE protein may act on Aβ keeping it soluble and preventing its aggregation, and ApoE4 variant may reduce Aβ solubility and increase aggregation in the brain. It is not fully understood at this time, what other proteins that might regulate Aβ solubility and prevent its aggregation. Understanding the endogenous mechanism of suppressing Aβ aggregation and enhancing its removal will help to target Aβ for developing disease-modifying drugs.
The neurotrophin receptor p75NTR may be such the protein which plays critical roles in the Aβ solubility and prevents Aβ aggregation and deposition in the brain. During the investigation into the functions of p75NTR in the development of AD in a recent study, we have found that the extracellular domain of p75NTR regulates the deposition of Aβ in a mouse model of AD.Citation7 In p75NTR gene-knockout APPswe/PS1dE9 mice, soluble Aβ which reflects the steady-state level of Aβ production, is reduced in the brain. The serum Aβ level, which is associated with the level of soluble Aβ in the brain, is also reduced in p75NTR-knockout animals. In comparison, we found that p75NTR knockout increases the insoluble Aβ as reflected by the increased amyloid plaques and formic acid-extracted Aβ levels. Our results indicate that p75NTR may play critical roles in the solubility of Aβ in the brain of AD mice. To test the hypothesis, we have made recombinant extracellular domain-fused with human immunoglobulin Fc fragment and tested its effects in the solubility of Aβ in vitro. We have found that the recombinant extracellular domain of p75NTR has a very strong effect on the solubility of Aβ. It reduces Aβ oligomerization and fibrillization, solubilizes fibrilized Aβ. Most interestingly, when injected into the hippocampus of AD mice, it reduces the number and size of Aβ plaques. Thus, we have clearly demonstrated that the extracellular domain plays an important role in the solubility of Aβ and might be one of endogenous mechanisms in the regulation of Aβ plaque formation in patients.
How might p75NTR play a role in the Aβ plaque formation or deposition in AD brain? One of the features of the Aβ pathology is that Aβ exclusively deposits in the neocortex, hippocampus and vessel walls. These areas are also the projection area of p75NTR positive fibers. The close anatomical association between p75NTR expression and Aβ deposition strongly suggests that p75NTR is involved in the initiation and development of Aβ deposition in the brain. Interestingly, we have observed there is a spatial relationship between p75NTR fibers and Aβ plaques in the brain of AD mice. We have found that p75NTR positive neurites locates in the center of compact senile plaques, while p75NTR negative degenerative neurites locate in the outer region of Aβ plaques.Citation7 This phenomenon suggests that p75NTR positive neurodegenerative fibers may play a seeding role to initiate the Aβ aggregation and plaque formation. It is known that Aβ can bind to the extracellular domain of p75NTR.Citation8 Normally, Aβ-bound p75NTR is likely endocytosed and degraded in the lysosomes, but the degenerated neurites may be abnormal in the endocytosis of the Aβ-p75NTR complex. Thus Aβ which binds to p75NTR on the cell surface may act as seeds to initiate the cascade of Aβ aggregation and beta-sheet formation.
On the other hand, the extracellular domain of p75NTR after enzymatic shedding may play a different role than the membranous p75NTR. It is known that the p75NTR extracellular domain is physiologically shedded by TACE to generate a soluble and diffusible factor.Citation9 The physiological function of the shedded diffusible extracellular domain of p75NTR remains unknown. During the aging process and during the development of AD, p75NTR expression is upregulated,Citation10,Citation11 and presumably the production of the diffusible extracellar domain of p75NTR is also increased. The increased extracellular domain of the p75NTR is likely a critical factor to maintain the solubility of Aβ, acting in concert with ApoE and other Aβ-binding proteins such as low-density lipoprotein receptor-related protein-1 (LRP1).Citation12 Indeed, in our animal experiment, we have found that knockout of p75NTR significantly increases the insoluble Aβ, even though the production of Aβ is reduced in p75NTR knockout neurons.Citation7 Our data have provided strong evidence that the brain Aβ deposition and amyloid plaque formation may be mainly due to the decreased Aβ solubility or decreased Aβ clearance.
The solubility of Aβ goes hand in hand with the clearance of Aβ because only the solubilized Aβ can be endocytosed and degraded in lysosomes by neurons, microglia and astrocytes, and be transported from the brain to the blood for degradation and clearance.Citation4 Whether p75NTR plays any roles in the endocytosis of Aβ and degradation is unclear. Our data of increased Aβ deposition in the brain of p75NTR knockout mice may also be explained by the decreased endocytosis of Aβ via p75NTR. It is likely that p75NTR may play a role in Aβ endocytosis, as p75NTR is a receptor of Aβ and mediates its toxicity in neurons.Citation8 p75NTR ligands can trigger a clathrin-dependent endocytosis of both p75NTR and its ligands.Citation13 If p75NTR normally mediates Aβ endocytosis, the knockout of p75NTR would reduce the removal of Aβ by endocytosis and lead to the increased deposition in the brain.
Normally, p75NTR is also expressed in endothelial cells and smooth muscle cells of blood vessels, vessel-innervating sympathetic and sensory neurons and choroid plexus in the brain.Citation14,Citation15 This raises the possibility that p75NTR within blood vessels may play a role in transport and trafficking of Aβ from the brain to the blood. It is well known that LRP1 plays important roles in the transport and transcytosis of Aβ and clears Aβ from the brain.Citation12 LRP1 on the cell-surface of the blood-brain barrier (BBB) can bind, transcytose and transport Aβ from the brain to the blood. p75NTR expressed on the BBB may play similar roles in the removal of Aβ in a similar manner to the Aβ binding proteins, LRP1 and G-glycoprotein, expressed on the BBB. Future studies should test roles of p75NTR in the endocytosis, transcytosis and clearance of Aβ by different cells such as neurons, endothelial cells and smooth muscle cells. The levels of shedded diffusible p75NTR in the brain and blood of AD patients should be determined as a biomarker and correlated with Aβ levels or Aβ plaques to reveal its potential roles in the solubility and clearance of Aβ in AD patients.
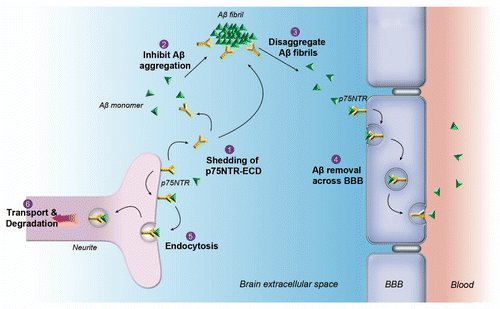
In summary, our studies have provided strong evidence that p75NTR is an essential molecule to keep the solubility of Aβ during development of AD. We speculate that p75NTR might also play many vital roles in removing Aβ from brain (). However, it is still far from certain how p75NTR regulates the solubility of Aβ and suppresses its deposition in the AD brain. Understanding the roles of p75NTR in the solubility and clearance of Aβ may help using p75NTR as a target to develop therapeutic drugs for the prevention and treatment of AD.
Figures and Tables
Figure 1 Schematic diagram depicting functions of p75NTR in Aβ solubility and clearance. p75NTR locates on the neurites, epithelial cells and smooth muscle cells of the blood-brain barrier (BBB). Binding of Aβ to p75NTR on neurites may initiate the endocytosis of Aβ and its degradation in the neurons. Shedding of p75NTR from the cell membrane releases the soluble extracellular domain (p75NTR-ECD), which is capable of inhibiting Aβ aggregation and disaggregating preformed Aβ fibrils. p75NTR at BBB might be able to transport Aβ from the brain to blood.
Acknowledgments
This work is supported by NHMRC grant No. 480422, FMC grants, NSFC grant No. 30973144 and Natural Science Foundation Project of CQCSTC No. CSTC2010BA5004. The authors thank Mr. Long Wei at Daping Hospital of Third Military Medical University for drawing the figure.
References
- Mawuenyega KG, Sigurdson W, Ovod V, Munsell L, Kasten T, Morris JC, et al. Decreased clearance of CNS beta-amyloid in Alzheimer's disease. Science 2010; 330:1774
- Selkoe DJ. Alzheimer's disease: genes, proteins and therapy. Physiol Rev 2001; 81:741 - 766
- Hardy JA, Higgins GA. Alzheimer's disease: the amyloid cascade hypothesis. Science 1992; 256:184 - 185
- Wang YJ, Zhou HD, Zhou XF. Clearance of amyloid-beta in Alzheimer's disease: progress, problems and perspectives. Drug Discov Today 2006; 11:931 - 938
- Blennow K, Zetterberg H. Cerebrospinal fluid biomarkers for Alzheimer's disease. J Alzheimers Dis 2009; 18:413 - 417
- Kim J, Basak JM, Holtzman DM. The role of apolipoprotein E in Alzheimer's disease. Neuron 2009; 63:287 - 303
- Wang YJ, Wang X, Lu JJ, Li QX, Gao CY, Liu XH, et al. p75NTR regulates Abeta deposition by increasing Abeta production but inhibiting Abeta aggregation with its extracellular domain. J Neurosci 2011; 31:2292 - 2304
- Yaar M, Zhai S, Pilch PF, Doyle SM, Eisenhauer PB, Fine RE, et al. Binding of beta-amyloid to the p75 neurotrophin receptor induces apoptosis. A possible mechanism for Alzheimer's disease. J Clin Invest 1997; 100:2333 - 2340
- Weskamp G, Schlondorff J, Lum L, Becherer JD, Kim TW, Saftig P, et al. Evidence for a critical role of the tumor necrosis factor alpha convertase (TACE) in ectodomain shedding of the p75 neurotrophin receptor (p75NTR). J Biol Chem 2004; 279:4241 - 4249
- Chakravarthy B, Gaudet C, Menard M, Atkinson T, Brown L, Laferla FM, et al. Amyloid-beta peptides stimulate the expression of the p75NTR neurotrophin receptor in SHSY5Y human neuroblastoma cells and AD transgenic mice. J Alzheimers Dis 2010; 19:915 - 925
- Costantini C, Weindruch R, Della Valle G, Puglielli L. A TrkA to p75NTR molecular switch activates amyloid beta-peptide generation during aging. Biochem J 2005; 391:59 - 67
- Shibata M, Yamada S, Kumar SR, Calero M, Bading J, Frangione B, et al. Clearance of Alzheimer's amyloid-ss(1-40) peptide from brain by LDL receptor-related protein-1 at the blood-brain barrier. J Clin Invest 2000; 106:1489 - 1499
- Bronfman FC, Tcherpakov M, Jovin TM, Fainzilber M. Ligand-induced internalization of the p75 neurotrophin receptor: a slow route to the signaling endosome. J Neurosci 2003; 23:3209 - 3220
- Spuch C, Carro E. The p75 neurotrophin receptor localization in blood-CSF barrier: expression in choroid plexus epithelium. BMC Neurosci 2011; 12:39
- Copray S, Kust B, Emmer B, Lin MY, Liem R, Amor S, et al. Deficient p75 low-affinity neurotrophin receptor expression exacerbates experimental allergic encephalomyelitis in C57/BL6 mice. J Neuroimmunol 2004; 148:41 - 53