Abstract
Prion diseases comprise a family of fatal neurodegenerative disorders caused by the conformational re-arrangement of a normal host-encoded protein, PrPC, to an abnormal infectious isoform termed PrPSc. Currently, the precise cellular mechanism(s) underlying prion disease pathogenesis remain unclear. Evidence suggests a role for the ubiquitin proteasome system (UPS), a protein degradation pathway that is critical for maintaining cellular proteostasis. Dysfunction of the UPS has been implicated in various neurodegenerative diseases. However, the mechanisms of this impairment remain unknown in many cases, and evidence that disease-associated misfolded proteins are able to directly inhibit the function of the proteasome has been lacking. Recently, we have shown data describing a mechanism of proteasome impairment by the direct interaction of β-sheet-rich PrP to reduce gate opening and inhibit substrate entry. This novel mechanism may provide a model for how other misfolded, disease-associated proteins might interact with the proteasome to disrupt its function. Targeting the UPS to restore proteostasis in neurodegenerative disorders in which misfolded proteins accumulate offers a possible target for therapeutic intervention.
Prion diseases are associated with the conversion of cellular prion protein (PrPC) to toxic β-sheet-rich isoforms (PrPSc) that are associated with disease pathogenesis.Citation1 Their neuropathology is characterized by tissue spongiosis, astrogliosis and extensive neuronal loss. To date, however, the exact cause(s) and underlying cellular events of prion-mediated neurodegeneration are poorly understood. While PrPC is critical for disease-associated PrP conversion to take place,Citation2 its loss is not the prime cause of disease.Citation3 Therefore, prion disease pathogenesis must be caused by a toxic gain-of-function resulting from the actual PrP conversion event. The conversion of PrPC to PrPSc in cells following prion infection is rapid, resulting in synthesis of PrPSc at the cell plasma membrane.Citation4 However, the means by which misfolded PrP traffics around the cell and its sub-cellular interactions remain ill-defined. Nevertheless, the PrP conversion process is associated with various alterations in cellular function, including signaling, metabolism, gene expression, and protein sorting and degradation.
Proteostasis is a key homeostatic process by which the amount and localization of proteins in the cell is regulated. Thus, all cells are able to selectively degrade damaged or redundant proteins, which could if they accumulated interfere with normal cellular function. One of the most important cellular mechanisms for maintaining proteostasis is the ubiquitin proteasome system (UPS). This pathway catalyzes the rapid elimination of misfolded proteins, and regulates the levels of many short-lived regulatory proteins associated with cellular metabolism and gene expression. It comprises a multi-step process by which proteins are tagged for rapid degradation by linkage to a chain of ubiquitin molecules, which act to target the substrate for hydrolysis by the 26S proteasome.Citation5 These large proteolytic complexes, resident in both the cytosol and nucleus of eukaryotic cells, consist of a 20S core particle and up to two 19S regulatory particles. Protein substrates targeted for degradation by the covalent linkage of a poly-ubiquitin chain are bound by the 19S particle, which disassembles the poly-ubiquitin chain and recycles the individual ubiquitin molecules. The substrate then undergoes an ATP-dependent process by which it is unfolded by the 19S particle and translocated through a narrow gated entry channel into the hollow cylindrical core of the 20S proteasome. Once inside this particle, the protein is cleaved into small peptides, which are released from the proteasome to be rapidly degraded by cytosolic peptidases.
Neurodegenerative diseases such as prion diseases, Alzheimer disease, Parkinson disease, Huntington disease and amyotrophic lateral sclerosis have hallmarks that differ from one another, but each is characterized by the accumulation and aggregation of specific misfolded proteins. These aggregated proteins may form inclusions such as Lewy bodies and neurofibrillary tangles, which are generally characterized by the presence of ubiquitin. Indeed, ubiquitin is a good, albeit non-specific, marker for many of the intracellular inclusions and neuropil deposits found in neurodegenerative disease. The UPS, therefore, could be viewed as a pathway that protects against neurodegenerative disease, impairment of which during on-going neurodegeneration may contribute to disease pathogenesis.Citation6 It is often proposed that the UPS is usually able to eliminate disease-associated misfolded proteins, but under pathogenic conditions such proteins gradually accumulate and aggregate to an extent that overwhelms the proteolytic capacity of a cell and impairs normal function of the UPS. Such a failure of the cellular proteolytic machinery could then cause an accumulation of other misfolded or abnormal proteins, leading to a breakdown in normal neuronal function. Indeed, conditional depletion in mice of 26S proteasomes in neurons of the substantia nigra or forebrain leads to neurodegeneration with inclusions resembling Lewy bodies.Citation7 However, evidence that the misfolded proteins in these various neurodegenerative diseases are able to directly inhibit the function of the proteasome has been lacking.
In prion diseases, the brains of mice infected with prions have increased levels of ubiquitin conjugates, which correlate with decreased proteasome function.Citation8 Neuronal cells infected with prions have reduced proteasomal activity against model peptide and protein substrates.Citation9 Moreover, cultured neuronal cells and transgenic mice have a decreased capacity to degrade ubiquitin-tagged fluorescent reporter proteins when infected with prions. Such observations are consistent with a loss of proteasomal activity, but could also be due to defects in other parts of the UPS pathway, such as ubiquitination. However, experiments using purified 26S proteasomes show a clear reduction in activity when they are incubated with recombinant PrP that is folded into a predominantly β-sheet-rich form similar to PrPSc, whereas a similar reduction is not observed when the PrP is folded into the α-helical structure of native PrPC.Citation9 This suggests that the changes in UPS function observed in prion disease are mediated, at least in part, by the direct actions of disease-associated PrP on the 26S proteasome.
Until recently, however, it remained unclear as to how exactly misfolded β-sheet-rich PrP inhibited the degradation of substrates by the proteasome. The 20S proteasome is a hollow barrel-shaped protein complex consisting of four stacked rings, each containing seven sub-units (). The two inner rings each comprise seven β-subunits, which together contain the six proteolytic active sites. These active sites differ in their specificity toward peptide bonds in protein and peptide substrates: two are chymotrypsin-like and cleave after hydrophobic amino acid residues; two are trypsin-like and cleave after basic residues; and two are caspase-like sites and cleave after acidic residues. Each of these proteolytic activities is reduced when proteasomes are exposed to β-sheet-rich PrP.Citation9 The two outer rings of the 20S particle contain seven α-subunits, the N-termini of which interact to form a gate that regulates substrate entry.Citation10 This gate is normally able to block substrate entry, and is usually opened by the six ATPase subunits (termed Rpt1–6) that comprise the base of the 19S regulatory particle, leading to increased substrate entry and hydrolysis. Therefore, for misfolded PrP aggregates to be able to directly inhibit the proteolytic sites of the proteasome, they would have to traverse both the ATPase ring of the 19S regulator and the even narrower (13 Å) gated pore of the 20S particle. This is unlikely given the likely size of the PrP aggregates and their probable resistance to disassembly by the 19S regulator. Thus, the most likely scenario seemed to be that disease-associated PrP aggregates act as a sticky plug that binds to the proteasome to prevent entry of other substrates into the 20S particle.
We tested this model, however, and found that the inhibitory effects of β-sheet-rich PrP on the 26S proteasome do not occur by the PrP acting as a plug to prevent substrate entry. Rather, the effect occurs via a novel mechanism of antagonism of proteasome gate opening and subsequent substrate entry.Citation11 This was demonstrated by examining the ability of β-sheet-rich PrP species to inhibit variants of the 20S and 26S proteasome that contain a constitutively open gate, allowing free access of substrates into the proteolytic core. Such constitutively open 20S particles are generated by a nine residue truncation of the α3-subunit N-terminus (α3ΔN) that prevents the formation of the closed gate conformation. These α3ΔN open-gated mutant proteasomes are readily able to degrade substrates in the absence of the activating 19S ATPases and have a much higher basal proteolytic activity than wild-type 20S proteasomes. Proteasome inhibitors that block the proteolytic sites readily inhibit α3ΔN open-gated mutant 20S proteasomes in the same way they do their wild-type counterparts. The same would be true for any agent acting as a sticky plug to block substrate entry into the 20S proteasome catalytic core. In contrast, however, if an agent directly affects the gate opening mechanism, then it should not be able to influence the peptidase activity of α3ΔN open-gated mutant proteasomes. This is exactly what was observed when β-sheet-rich PrP was incubated with either α3ΔN open-gated 20S or 26S particles, in contrast to the inhibitory actions seen in wild-type proteasomes.Citation11 These data suggest that disease-associated misfolded PrP isoforms inhibit gate opening in the 20S proteasome, which accounts for the inhibition of 26S proteasomal function.
The induction of 20S proteasome gate opening by the 19S regulator is regulated by two specific ATPase subunits, Rpt1 and Rpt5. These subunits bind ATP and then dock via their C-termini into intersubunit pockets formed by the α-subunits of the 20S particle in a ‘key-in-a-lock’ fashion to stimulate gate opening and substrate entry.Citation12 This process can be mimicked using a synthetic 8-residue peptide corresponding to the C-terminus of the Rpt5 ATPase, termed CtRpt5, addition of which is sufficient to induce 20S proteasome gate opening and substrate hydrolysis. Misfolded PrP aggregates are able to reduce substrate hydrolysis by CtRpt5-activated 20S proteasomes as they do the 26S proteasome complex.Citation11 However, in the presence of β-sheet-rich PrP, the CtRpt5 peptide is still able to induce some increase in 20S proteasome gate opening. Thus, rather than directly blocking the actions of CtRpt5, it appears that disease-associated misfolded PrP conformers enhance the closed state of the 20S proteasome gate, which in the presence of the gate-opening properties of CtRpt5 results in a partial gate opening response.
Gate opening in the 20S proteasome can also be stimulated by another family of regulatory particles, the 11S activators. These regulators, like the 19S regulator, bind to the ends of the 20S proteasome and induce gate opening, but they do so by a mechanism that is different to the 19S ATPases. It is intriguing, therefore, that disease-associated PrP conformers are not able to reduce the activity of 11S-activated 20S proteasomes, in stark contrast to their effect on 20S proteasome gate opening by the 19S ATPases (or their mimetic, the CtRpt5 peptide).Citation11 The 11S activators associate with the 20S proteasome by docking into the same intersubunit pockets of the α-ring as the 19S ATPases.Citation13 However, unlike the 19S ATPases, this is not sufficient for the 11S activators to stimulate 20S proteasome gate opening, requiring instead a separate activation domain. Therefore, the means by which the 20S proteasome gate is opened by either the 19S ATPases or 11S activators appears to be quite different. Indeed, while the 11S activators induce gate opening without a major conformational change to the 20S proteasome α-subunits,Citation14 the 19S ATPases do so with a large 4° rotation of the entire α-ring.Citation15 It is conceivable, therefore, that β-sheet-rich PrP acts to inhibit the 20S proteasome by stabilizing its un-rotated, closed-gate conformation, which would reduce gate opening by the 19S ATPases, but not gate opening without α-subunit rotation by the 11S activators.
Taken together, these observations raise an obvious question—how do disease-associated misfolded PrP conformers bind to and inhibit the 20S proteasome? Despite the fact that 11S-activated complexes are not inhibited, β-sheet-rich PrP remains bound to the 20S proteasome in the presence of saturating concentrations of the 11S activator, PA26.Citation11 Given that these PrP aggregates are too large to enter the 20S proteasome pore, and that PA26 binds tightly to the α-subunits that form the ends of the 20S proteasome barrel, then by a process of exclusion it can be concluded that β-sheet-rich PrP must bind to the outer lateral surface of the 20S proteasome (). This explains the inhibitory effect on 26S proteasomes of these misfolded PrP aggregates without any displacement of the 19S regulatory particles,Citation9 which bind to the same end surface of the 20S proteasome as PA26. This association with the lateral surface of the 20S particle probably interferes with the rotation of the α-subunits that is induced by the 19S ATPases, but has no effect on the ability of 11S activators to cause gate opening via their activation loop without α-subunit rotation. While a location somewhere near the α-ring is most probable, the exact site of β-sheet-rich PrP binding to the 20S proteasome remains to be elucidated. Further structural data would be informative, but obtaining such information is difficult because of the heterogeneous and insoluble nature of the protein aggregates involved.
The exact nature of the inhibitory PrP species that interact with the 20S proteasome also remains to be determined. Structural studies of recombinant PrP molecules folded into a β-sheet-rich conformation demonstrate a protease resistant core that can either form insoluble amyloid fibrils or soluble prefibrillar oligomers.Citation16,Citation17 The precise size and structure of such oligomers remain poorly defined, but PrP oligomers have been implicated in prion disease toxicity and infectivity. In relation to the inhibitory effects of β-sheet-rich PrP species on the proteasome, they are abrogated by pre-incubating such PrP preparations with an antibody raised against protein aggregation intermediates, suggesting that they are oligomeric.Citation9 Moreover, PrP amyloid fibrils do not inhibit the proteolytic activity of the 26S proteasome. The apparent IC50 of the actions of aggregated recombinant β-sheet-rich PrP on 20S proteasomes is 90–180 nM monomeric PrP.Citation11 This corresponds to a molar ratio of 10–20 monomeric PrP molecules per 20S proteasome in solution, but the concentration of PrP aggregates must be much lower (although the mean number of PrP monomers per inhibitory aggregate remains unknown). For the moment, however, precise structural characterization of the inhibitory PrP species of the 20S proteasome is not possible, again due to the heterogeneous and insoluble character of PrP aggregates.
Although it is now clear that β-sheet-rich PrP species can interfere with the gating mechanism of the proteasome, the degree to which this contributes to the evident defect in protein degradation in prion-infected cells remains unknown. For example, it is possible that in addition to its direct actions on the proteasome, aggregating PrP may also sequester components of the UPS such that its degradative capacity becomes compromised, or such species may perhaps exert other, indirect effects on the UPS via altered cellular metabolism and caspase activation. Moreover, the degree to which dysfunction of the UPS per se is a critical toxic mechanism in prion diseases remains unknown. Recent findings that transgenic mice expressing mutant, albeit non-infectious, forms of PrP associated with certain inherited human prion diseases do not show UPS impairment,Citation18 contrast with the clear accumulation of ubiquitinated proteins and reporter substrates indicating UPS dysfunction in prion-infected cells and animals.Citation8,Citation9 Certainly, prion disease pathogenesis is likely to be multi-factorial, but the critical nature of the UPS in serving as a quality-control system to maintain cellular proteostasis makes it easy to envisage that a loss of proteasome function could contribute to the perturbation and loss of neurons characteristic of disease pathogenesis. Indeed, it is unsurprising, perhaps, that the addition of proteasome inhibitors invariably promotes increased neuronal dysfunction and death in cellular models of neurodegenerative diseases. Moreover, several short-lived proteins known to be UPS substrates have been shown to accumulate in the brains of prion-infected mice.Citation11 Further studies to determine how early in disease the UPS dysfunction occurs will go some way to establishing its likely importance in prion disease pathogenesis.
The finding that β-sheet-rich PrP impairs the UPS by interacting with the 20S proteasome to antagonize gate opening and inhibit substrate entry serves as a model for how other misfolded proteins associated with neurodegeneration may impair UPS function. For example, in Alzheimer disease, where transgenic mouse models show impaired proteasome activity that correlates with accumulating amyloid β-protein (Aβ) oligomers,Citation19,Citation20 in vitro studies have shown that Aβ oligomers inhibit 20S proteasomes to reduce their proteolytic activity.Citation20,Citation21 In Huntington disease, UPS dysfunction is observed following the induction of protein aggregation and inclusion formation by mutant huntingtin.Citation22 However, while global changes in the cellular ubiquitin system are observed in HD, with the lysine-48-linked poly-ubiquitin chains associated with targeting substrates to the proteasome being shown to accumulate early in disease pathogenesis,Citation23 this appears in at least one transgenic mouse model to be in the absence of any detectable impairment of proteasome catalytic function.Citation24 In a mouse model of another polyglutamine disease, spinocerebellar ataxia 7, pathogenesis occurs in the absence of any detectable UPS impairment.Citation25 In Parkinson disease, it has been shown that α-synuclein oligomers co-precipitate with the 26S proteasome and inhibit proteasomal activity,Citation26,Citation27 although the mechanism of this inhibition has not been elucidated. Therefore, it is feasible that antagonistic actions on the 20S proteasome, such as those by b-sheet-rich PrP, may account for the UPS dysfunction observed in other neurodegenerative diseases. Given the recent description of a selective small-molecule inhibitor of the human proteasome-associated deubiquitinating enzyme, USP14, which enhances proteasome activity and the degradation of tau, ataxin-3 and TDP-43 in cultured cells,Citation28 enhancing proteasomal activity may offer a means of reducing the levels of misfolded proteins associated with neurodegenerative diseases.
Figures and Tables
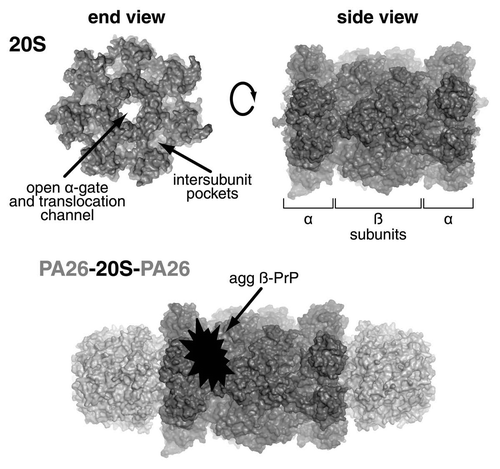
Figure 1 The 20S proteasome comprises four rings of seven subunits each. The two inner rings each contain seven β-subunits, including the β1, β2 and β5 sites that exhibit caspase-like, trypsin-like and chymotrypsin-like activities respectively. The two outer rings each contain seven α-subunits, the N-termini of which form the gate that prevents substrate entry in its closed state, but when open allows substrate entry through the translocation pore into the catalytic core. The 19S ATPases dock into the intersubunit pockets of the α-ring to cause gate opening. Similarly, 11S activators, such as the PA26 shown here, also bind to the α-ring intersubunit pockets, but cause gate opening by an alternative mechanism. The finding that aggregated β-sheet-rich PrP and PA26 do not bind to the same part of the 20S proteasome illustrates that such PrP species cause their antagonistic effect on gate opening by binding to the proteasome's lateral surface. Figure adapted from Deriziotis P, et al. EMBO J 2011; 30:3065–77, courtesy of David M. Smith, West Virginia University School of Medicine.
Acknowledgments
We are grateful to Pelagia Deriziotis, David Smith and Alfred Goldberg for discussion and comment, and to Ray Young for preparation of graphics. This work in our laboratory has been supported by the UK Medical Research Council, the UK Department of Health and the Brain Research Trust.
References
- Prusiner SB. Novel proteinaceous infectious particles cause scrapie. Science 1982; 216:136 - 144; PMID: 6801762; http://dx.doi.org/10.1126/science.6801762
- Büeler H, Aguzzi A, Sailer A, Greiner RA, Autenried P, Aguet M, et al. Mice devoid of PrP are resistant to scrapie. Cell 1993; 73:1339 - 1347; PMID: 8100741; http://dx.doi.org/10.1016/0092-8674(93)90360-3
- Mallucci GR, Ratte S, Asante EA, Linehan J, Gowland I, Jefferys JG, et al. Post-natal knockout of prion protein alters hippocampal CA1 properties, but does not result in neurodegeneration. EMBO J 2002; 21:202 - 210; PMID: 11823413; http://dx.doi.org/10.1093/emboj/21.3.202
- Goold R, Rabbanian S, Sutton L, Andre R, Arora P, Moonga J, et al. Rapid cell-surface prion protein conversion revealed using a novel cell system. Nat Commun 2011; 2:281; PMID: 21505437; http://dx.doi.org/10.1038/ncomms1282
- Glickman MH, Ciechanover A. The ubiquitin-proteasome proteolytic pathway: destruction for the sake of construction. Physiol Rev 2002; 82:373 - 428; PMID: 11917093
- Rubinsztein DC. The roles of intracellular protein-degradation pathways in neurodegeneration. Nature 2006; 443:780 - 786; PMID: 17051204; http://dx.doi.org/10.1038/nature05291
- Bedford L, Hay D, Devoy A, Paine S, Powe DG, Seth R, et al. Depletion of 26S proteasomes in mouse brain neurons causes neurodegeneration and Lewy-like inclusions resembling human pale bodies. J Neurosci 2008; 28:8189 - 8198; PMID: 18701681; http://dx.doi.org/10.1523/JNEUROSCI.2218-08.2008
- Kang SC, Brown DR, Whiteman M, Li R, Pan T, Perry G, et al. Prion protein is ubiquitinated after developing protease resistance in the brains of scrapie-infected mice. J Pathol 2004; 203:603 - 608; PMID: 15095484; http://dx.doi.org/10.1002/path.1555
- Kristiansen M, Deriziotis P, Dimcheff DE, Jackson GS, Ovaa H, Naumann H, et al. Disease-associated prion protein oligomers inhibit the 26S proteasome. Mol Cell 2007; 26:175 - 188; PMID: 17466621; http://dx.doi.org/10.1016/j.molcel.2007.04.001
- Groll M, Bajorek M, Kohler A, Moroder L, Rubin DM, Huber R, et al. A gated channel into the proteasome core particle. Nat Struct Biol 2000; 7:1062 - 1067; PMID: 11062564; http://dx.doi.org/10.1038/80992
- Deriziotis P, Andre R, Smith DM, Goold R, Kinghorn KJ, Kristiansen M, et al. Misfolded PrP impairs the UPS by interaction with the 20S proteasome and inhibition of substrate entry. EMBO J 2011; 30:3065 - 3077; PMID: 21743439; http://dx.doi.org/10.1038/emboj.2011.224
- Smith DM, Chang SC, Park S, Finley D, Cheng Y, Goldberg AL. Docking of the proteasomal ATPases' carboxyl termini in the 20S proteasome's alpha ring opens the gate for substrate entry. Mol Cell 2007; 27:731 - 744; PMID: 17803938; http://dx.doi.org/10.1016/j.molcel.2007.06.033
- Whitby FG, Masters EI, Kramer L, Knowlton JR, Yao Y, Wang CC, et al. Structural basis for the activation of 20S proteasomes by 11S regulators. Nature 2000; 408:115 - 120; PMID: 11081519; http://dx.doi.org/10.1038/35040607
- Förster A, Masters EI, Whitby FG, Robinson H, Hill CP. The 1.9 Å structure of a proteasome-11S activator complex and implications for proteasome-PAN/PA700 interactions. Mol Cell 2005; 18:589 - 599; PMID: 15916965; http://dx.doi.org/10.1016/j.molcel.2005.04.016
- Yu Y, Smith DM, Kim HM, Rodriguez V, Goldberg AL, Cheng Y. Interactions of PAN's C-termini with archaeal 20S proteasome and implications for the eukaryotic proteasome-ATPase interactions. EMBO J 2010; 29:692 - 702; PMID: 20019667; http://dx.doi.org/10.1038/emboj.2009.382
- Martins SM, Frosoni DJ, Martinez AM, De Felice FG, Ferreira ST. Formation of soluble oligomers and amyloid fibrils with physical properties of the scrapie isoform of the prion protein from the C-terminal domain of recombinant murine prion protein mPrP-(121–231). J Biol Chem 2006; 281:26121 - 26128; PMID: 16844683; http://dx.doi.org/10.1074/jbc.M605367200
- Gerber R, Tahiri-Alaoui A, Hore PJ, James W. Oligomerization of the human prion protein proceeds via a molten globule intermediate. J Biol Chem 2007; 282:6300 - 6307; PMID: 17210575; http://dx.doi.org/10.1074/jbc.M608926200
- Quaglio E, Restelli E, Garofoli A, Dossena S, De Luigi A, Tagliavacca L, et al. Expression of mutant or cytosolic PrP in transgenic mice and cells is not associated with endoplasmic reticulum stress or proteasome dysfunction. PLoS ONE 2011; 6:19339; PMID: 21559407; http://dx.doi.org/10.1371/journal.pone.0019339
- Keller JN, Hanni KB, Markesbery WR. Impaired proteasome function in Alzheimer's disease. J Neurochem 2000; 75:436 - 439; PMID: 10854289; http://dx.doi.org/10.1046/j.1471-4159.2000.0750436.x
- Tseng BP, Green KN, Chan JL, Blurton-Jones M, LaFerla FM. Abeta inhibits the proteasome and enhances amyloid and tau accumulation. Neurobiol Aging 2008; 29:1607 - 1618; PMID: 17544172; http://dx.doi.org/10.1016/j.neurobiolaging.2007.04.014
- Gregori L, Fuchs C, Figueiredo-Pereira ME, Van Nostrand WE, Goldgaber D. Amyloid beta-protein inhibits ubiquitin-dependent protein degradation in vitro. J Biol Chem 1995; 270:19702 - 19708; PMID: 7649980; http://dx.doi.org/10.1074/jbc.270.34.19702
- Bence NF, Sampat RM, Kopito RR. Impairment of the ubiquitin-proteasome system by protein aggregation. Science 2001; 292:1552 - 1555; PMID: 11375494; http://dx.doi.org/10.1126/science.292.5521.1552
- Bennett EJ, Shaler TA, Woodman B, Ryu KY, Zaitseva TS, Becker CH, et al. Global changes to the ubiquitin system in Huntington's disease. Nature 2007; 448:704 - 708; PMID: 17687326; http://dx.doi.org/10.1038/nature06022
- Bett JS, Goellner GM, Woodman B, Pratt G, Rechsteiner M, Bates GP. Proteasome impairment does not contribute to pathogenesis in R6/2 Huntington's disease mice: exclusion of proteasome activator REGgamma as a therapeutic target. Hum Mol Genet 2006; 15:33 - 44; PMID: 16311253; http://dx.doi.org/10.1093/hmg/ddi423
- Bowman AB, Yoo SY, Dantuma NP, Zoghbi HY. Neuronal dysfunction in a polyglutamine disease model occurs in the absence of ubiquitin-proteasome system impairment and inversely correlates with the degree of nuclear inclusion formation. Hum Mol Genet 2005; 14:679 - 691; PMID: 15661755; http://dx.doi.org/10.1093/hmg/ddi064
- Lindersson E, Beedholm R, Hojrup P, Moos T, Gai W, Hendil KB, et al. Proteasomal inhibition by alpha-synuclein filaments and oligomers. J Biol Chem 2004; 279:12924 - 12934; PMID: 14711827; http://dx.doi.org/10.1074/jbc.M306390200
- Emmanouilidou E, Stefanis L, Vekrellis K. Cell-produced alpha-synuclein oligomers are targeted to, and impair, the 26S proteasome. Neurobiol Aging 2010; 31:953 - 968; PMID: 18715677; http://dx.doi.org/10.1016/j.neurobiolaging.2008.07.008
- Lee BH, Lee MJ, Park S, Oh DC, Elsasser S, Chen PC, et al. Enhancement of proteasome activity by a small-molecule inhibitor of USP14. Nature 2010; 467:179 - 184; PMID: 20829789; http://dx.doi.org/10.1038/nature09299