Abstract
Prion diseases comprise a group of rapidly progressive and invariably fatal neurodegenerative disorders for which there are no effective treatments. While conversion of the cellular prion protein (PrPC) to a β-sheet rich isoform (PrPSc) is known to be a critical event in propagation of infectious prions, the identity of the neurotoxic form of PrP and its mechanism of action remain unclear. Insights into this mechanism have been provided by studying PrP molecules harboring deletions and point mutations in the conserved central region, encompassing residues 105–125. When expressed in transgenic mice, PrP deleted for these residues (Δ105–125) causes a spontaneous neurodegenerative illness that is reversed by co-expression of wild-type PrP. In cultured cells, Δ105–125 PrP confers hypersensitivity to certain cationic antibiotics and induces spontaneous ion channel activity that can be recorded by electrophysiological techniques. We have utilized these drug-hypersensitization and current-inducing activities to identify which PrP domains and subcellular locations are required for toxicity. We present an ion channel model for the toxicity of Δ105–125 PrP and related mutants and speculate how a similar mechanism could mediate PrPSc-associated toxicity. Therapeutic regimens designed to inhibit prion-induced toxicity, as well as formation of PrPSc, may prove to be the most clinically beneficial.
The Mechanism of Prion Toxicity is Unknown
Transmissible spongiform encephalopathies or prion diseases, comprise a unique group of neurodegenerative disorders in humans and animals that can be genetic, infectious or sporadic in origin.Citation1 There are currently no effective treatments or cures for prion diseases, and death typically occurs within several years of the onset of symptoms. There is now strong evidence that prion infectivity is propagated by conformational conversion of the normal, cellular prion protein (PrPC) to an aggregated, β-sheet-rich isoform (PrPSc).Citation2 Neuronal loss and synaptic dysfunction are major components of prion pathology, yet the underlying molecular mechanism of this neurotoxicity remains unclear. There is evidence that PrPSc itself is not the toxic isoform of PrP, since large amounts of PrPSc can accumulate without evident neuropathology,Citation3,Citation4 and prion-induced pathology can occur with very low levels of PrPSc.Citation5,Citation6 PrP knockout mice display only minor phenotypic abnormalities, and the physiological role of PrPC remains uncertain.Citation7 Thus, a simple loss of PrPC function cannot account for prion neurotoxicity.Citation8 Interestingly, the presence of PrPC has been shown to be critical, not only as a substrate for PrPSc propagation, but also as a transducer of PrPSc-associated neuronal death.Citation9 This second function of PrPC requires anchoring to the cell surface.Citation10 These observations suggest that the PrPC to PrPSc conversion process produces an intermediate or byproduct, most likely membrane anchored, that is responsible for prion toxicity, and that this toxic species may work by altering a normal, functional activity of PrPC.
In order to investigate the mechanisms of prion toxicity, we have studied a mutant form of PrP (referred to as ΔCR) that is missing amino acids 105–125, which lie in the highly conserved central region of the protein. Transgenic mice expressing ΔCR PrP display a spontaneous neurodegenerative illness that is lethal in the neonatal period on the PrP-null background, and is characterized by massive death of cerebellar granule neurons, as well as degeneration of white matter in the brain and spinal cord.Citation11 ΔCR PrP is not aggregated or protease resistant, and its pattern of cellular localization is similar to that of wild-type (WT) PrP.Citation11,Citation12 The ΔCR PrP neurodegenerative phenotype can be dose-dependently suppressed by co-expression of WT PrP, suggesting that the mutant and WT proteins may be acting in a similar pathway, and that ΔCR PrP might be toxic because it alters a normal functional activity of PrPC.Citation11 Since a similar subversion of PrPC function may be involved in the pathogenesis of infectious prion diseases, information gleaned from the ΔCR PrP model of toxicity could be useful in designing anti-prion therapeutics.
ΔCR PrP Induces Spontaneous Ionic Currents
We were initially surprised to find that ΔCR PrP expression in cultured cells resulted in minimal cell death, in contrast to the dramatic neuronal loss observed in transgenic mice that express this protein. Subsequently, we observed that ΔCR PrP-expressing cells were hypersensitive to certain cationic antibiotics (such as G418, hygromycin and Zeocin) that are typically used for selection of stably transfected cell clones. This phenomenon became the basis for a drug-based cellular assay (DBCA).Citation13 Related cationic drugs have been shown to enter cells down an electrochemical gradient through ion channels,Citation14,Citation15 and we observed that depolarizing the membrane potential by raising extracellular potassium levels completely suppressed drug-induced toxicity in cells expressing ΔCR PrP.Citation16 Therefore, we decided to examine the electrophysiological properties of ΔCR PrP-expressing cells by whole-cell patch clamping.
These experiments revealed large, spontaneous inward currents in transfected cells expressing ΔCR PrP ().Citation17 Through a series of ion substitution experiments, we determined that the currents were generated by a non-selective, cation-permeable channel with unique properties. These currents were observed in a variety of non-neuronal cell types, including HEK, CHO and Sf9 cells, which have few endogenous channels. Thus, we hypothesize that the spontaneous currents are generated by ion channels or pores in the plasma membrane formed by ΔCR PrP molecules themselves, without the intervention of endogenous cellular proteins. Similar currents were observed in cerebellar granule neurons from Tg(ΔCR) mice, both in dissociated culture and in brain slices (unpublished data).
In a recent study, we used both the DBCA and whole-cell patch clamping to identify regions of PrP required for ion channel activity.Citation18 A schematic of the murine prion protein with its domain structure is shown in . The main results of the structure-function study are summarized in . A standard set of experiments was conducted for each construct, which were stably expressed in HEK cells: western blotting to confirm expression levels and glycosylation patterns, immunofluorescence staining to determine cellular localization, treatment with G418 to measure cell death due to drug hypersensitivity, and whole-cell patch clamping to detect the generation of spontaneous inward currents.
The N-terminal, Polybasic Domain is Required for PrP Channel Activity
One of the striking findings of our study was that the N-terminal polybasic domain of PrP (residues 23–31; KKR PKP GGW) was required for ion channel activity as well as drug hypersensitivity. Thus, cells expressing Δ23–31/ΔCR PrP exhibited neither spontaneous currents nor significant G418-induced cell death.Citation18 Why is the 23–31 region so important? Some clues may be gleaned from the known cell biological functions associated with this region.
One proposed function is that of a protein transduction domain (PTD), the prototype of which is found in the HIV Tat protein.Citation19 PTDs are short, positively-charged sequences that are capable of ferrying passenger proteins across the lipid bilayer by mechanisms that are still not completely understood.Citation20 Residues 23–29 of PrP are homologous to the PTD of HIV Tat, and have been shown to be capable of transducing reporter proteins across the lipid bilayer.Citation19 We propose that the N-terminus of ΔCR PrP inserts into the plasma membrane by virtue of the PTD activity of residues 23–31. However, due to the presence of the glycosyl-phosphatidylinositol (GPI) anchor attaching the C-terminus to the plasma membrane, the protein is unable to completely translocate into the cytoplasm. This may result in transient formation of channels or pores in the plasma membrane (). The randomly fluctuating nature of ΔCR PrP-induced currents is consistent with this model.
A second function of residues 23–31 may also be relevant to ΔCR PrP activity. These residues constitute one of three glycosaminoglycan (GAG) binding domains in PrP,Citation21 and this region mediates interactions with glypican-1, a cell-surface proteoglycan.Citation22 Thus, deletion of residues 23–31 may reduce the ability of ΔCR PrP to interact with cellular GAGs, which may in turn impact channel formation. We have shown that treatment with exogenous GAGs such as pentosan polysulfate rapidly silences ΔCR PrP-induced currents, perhaps by competing with endogenous GAGs.Citation18 Of note, GAG binding is thought to be important for the action of PTDs.Citation20
A third documented activity of the N-terminal polybasic domain is to mediate endocytosis of PrP via clathrin-coated pits.Citation23,Citation24 However, it is unlikely that endocytosis is involved in ΔCR PrP activity, as we have found that point mutations in this region that eliminate endocytosis have no effect on drug hypersensitivity.Citation18
Channel Activity Requires Membrane Attachment
Another major finding of our study is that attachment to the plasma membrane, but not localization to lipid rafts, is critical for ion channel activity, as shown by a series of ΔCR PrP variants targeted to various cellular locations. PrPC, due to its GPI anchor, localizes to detergent-resistant membrane domains or lipid rafts.Citation25 Elimination of the GPI attachment signal causes the protein to be secreted into the medium, while replacement of this segment with a transmembrane domain results in cell surface attachment, but exclusion from lipid rafts.Citation26 Ion channel activity was eliminated in the anchorless form, but preserved in the transmembrane form of ΔCR PrP.Citation18 Membrane bound forms of ΔCR PrP that were targeted to the ER or Golgi also lacked channel activity.Citation18
Taken together, these findings indicate that ΔCR PrP must be attached to the plasma membrane to exert its current-inducing and drug-hypersensitizing activities, but that residence in lipid rafts is not required. The requirement for membrane attachment is consistent with the formation of ion channels on the cell surface by ΔCR PrP. If the N-terminal polybasic domain were acting as a tethered protein transduction domain as discussed above, then lack of membrane attachment would eliminate toxicity by allowing ΔCR PrP molecules to completely translocate to the cytoplasm, rather than transiently forming pores in the membrane.
Other Mutations in the Central Region Induce Ion Channel Activity
To understand why the deletion of residues in the unstructured central region of PrP induces such a strong toxic effect, we made several additional deletions corresponding to PrP constructs expressed in transgenic mice.Citation11,Citation27,Citation28 The deletion of residues 105–125 eliminates part of a cluster of positively charged amino acids as well as part of a hydrophobic domain. Additional deletions included Δ94–110 (all of the charged cluster), Δ111–134 (all of the hydrophobic domain), Δ114–121 (part of the hydrophobic domain), and Δ94–134 (the entire charged cluster and hydrophobic domain). As expected, Δ94–134 induced a similar effect as ΔCR PrP. Surprisingly, however, we found that the non-overlapping deletions Δ94–110 and Δ111–134 both produced toxic effects. This could be due to the disruption of a binding site requiring both the charged cluster and hydrophobic domain to be intact. Alternatively, deletion of amino acids in this region could alter the flexibility of the N-terminus of PrP, allowing it to interact with the membrane in an abnormal fashion. Consistent with this suggestion, binding of anti-PrP antibodies 6D11 and D13 to amino acids immediately upstream of the central region rescues toxicity.Citation13
We also examined the effect of mutations associated with familial prion diseases in humans (Creutzfeldt-Jakob disease and Gerstmann-Sträussler-Scheinker syndrome), focusing on those that lie within and near the central region that is deleted in ΔCR.Citation18 Of the nine point mutants we examined, only three (P101L, G113V and G130V, representing the mouse homologs of the human mutations) induced channel activity and drug hypersensitivity. It could be that the other mutants have activity that is below the threshold of detectability in these assays. In fact, there is a correlation between the activity of the mutants in our assays and their pathogenicity in mice and humans. Thus, Tg(P101L) mice get sick later than Tg(ΔCR) mice.Citation29 Similarly, the G114V mutation in humans, which has the greatest effect in our assays, is associated with an extremely early onset in human patients.Citation30 The three point mutants displaying activity exhibited smaller effects than the ΔCR mutant, which might be expected, given the more dramatic sequence change introduced by the deletion.
To investigate why some mutants had lower activity, we determined the level of aggregation of each mutant by assaying detergent insolubility, and the amount of cell surface expression using a cell-blotting assay.Citation18 There was no correlation between these two properties and the G418-hypersensitizing activities of the mutants. Mutations outside the central region, including a 9-octapeptide repeat insertion (PG14) and D177N, cause neurodegenerative phenotypes in humans and transgenic mice,Citation31,Citation32 but lacked detectable ion channel or drug hypersensitizing activities in our assays.
Taken together, our results suggest that ion channel hyperactivity may contribute to the pathogenesis of some, but not all inherited prion disorders.
Ion Channel Model for PrP Toxicity
Our recent studies demonstrate that PrP molecules carrying deletions or mutations in the central region induce spontaneous currents in transfected cells, and that this phenomenon requires cell surface localization of the PrP, as well as an intact, N-terminal polybasic domain. Our data support a model in which ΔCR PrP molecules themselves form ion channels or pores in the plasma membrane, possibly via the PTD activity of residues 23–31 (). Increased permeability to extracellular ions could then induce neuronal degeneration via an excitotoxic mechanism, either by inducing spontaneous electrical activity, or by sensitizing neurons to normal synaptic input. Consistent with this suggestion, we have found that granule cell neurons in Tg(ΔCR) mice degenerate by a distinctive, non-apoptotic mechanism that bears similarities to the pathways associated with glutamate-induced excitotoxicity.Citation33 Moreover, neurons expressing ΔCR PrP are more sensitive to the excitotoxic effects of exogenously applied glutamate (unpublished data). WT PrP rescues ΔCR PrP toxicity in a dose dependent manner, which might be explained by a homo-oligomerization process in which increasing proportions of WT PrP subunits dampen the effects of ΔCR PrP ().
The abnormal ion channel activity associated with some PrP point mutations may contribute to the pathogenesis of inherited forms of prion disease. Channel activity may also be relevant to the neurotoxic mechanisms initiated by infectious PrPSc. ΔCR PrP may resemble an intermediate or side product produced during conversion of PrPC to PrPSc (). Indeed, the central region of PrPC is known to undergo major conformation changes during formation of PrPSc.Citation34–Citation36 If this model were correct, compounds we have identified that silence ΔCR PrP-induced currents would be predicted to mitigate the disease process in both Tg(ΔCR) as well as scrapie-infected mice. Thus far, attempts to treat prion diseases by inhibiting the PrPC to PrPSc conversion process have had limited success in stopping or reversing pathology.Citation37 This may be due to the fact that toxic forms of PrP, once produced, remain active even if further conversion is inhibited. Therefore, the most effective anti-prion treatments could be those that block prion-induced toxic pathways, as well as PrPSc formation.
An ion channel model for PrP may have relevance to other neurodegenerative disorders. Recent reports suggest that PrPC is a receptor for oligomeric assemblies of the Alzheimer Aβ peptide, and possibly other β-rich proteins.Citation38,Citation39 Binding of these aggregates could induce an ion channel activity in PrPC similar to that caused by the ΔCR deletion (). Of note, the two binding sites on PrPC for Aβ oligomers lie in the N-terminal polybasic region (residues 23–28), and immediately adjacent to the central region (residues 95–105), domains that are known to affect the ion channel activity of PrP. Thus, PrP-associated ion channels may represent an attractive, new therapeutic target for prion diseases as well as other neurodegenerative disorders due to protein misfolding.
Figures and Tables
Figure 1 Spontaneous inward currents induced by expression of ΔCR PrP in HEK cells. HEK cells expressing vector, WT PrP, ΔCR PrP or ΔCR + WT PrP were analyzed by whole-cell patch-clamping. Recordings were made at a holding potential of −80 mV. Spontaneous current activity was observed only in cells expressing ΔCR PrP, and was suppressed by co-expression of WT PrP.
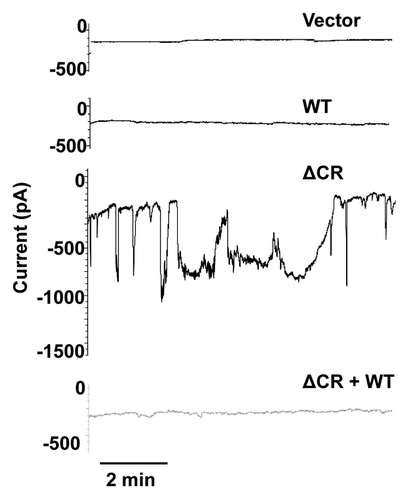
Figure 2 Schematic of murine PrP showing domain structure and mutations. ER signal sequence (SS, blue, aa 1–22); polybasic domain (yellow, aa 23–31); octapeptide repeats (OR, magenta, aa 51–90); charge cluster (CC, red, aa 94–110); hydrophobic domain (HD, green, aa 111–134); GPI signal sequence (GPI, purple, 231–254). Three α helices (H1, H2 and H3), two N-linked oligosaccharides (hexagons), and one disulfide bond (S-S) are present in the C-terminal half of PrP. The sequences for amino acids 23–31 and 94–134 are shown above the schematic. The boxed residues (105–125) indicate those deleted in ΔCR PrP. Residues mutated in familial prion diseases are indicated in red, and a nonpathogenic polymorphism in blue, with arrows pointing to the substituted amino acid.
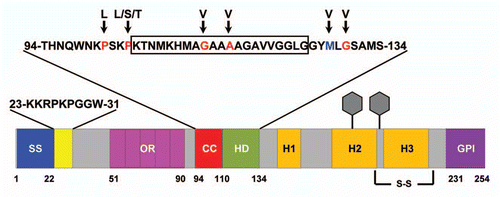
Figure 3 Ion Channel Formation Model of PrP-dependent toxicity. (A) ΔCR PrP, as a result of deletion of amino acids 105–125 (red), creates channels or pores in the plasma membrane by insertion of N-terminal, polybasic region (residues 23–31, shown in yellow). This allows cations (purple balls) to permeate the plasma membrane. The presence of WT PrP closes the channel. (B) PrPSc (orange) or oligomers of the Aβ peptide (red) bind to PrPC and induce a ΔCR-like conformational change, resulting in transient pore formation and toxicity.
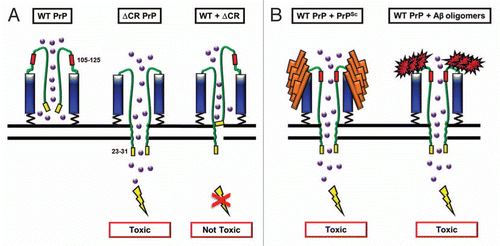
Table 1 Summary of structure-function study
Acknowledgments
Work in the Harris laboratory is supported by National Institutes of Health Grants NS052526 and NS040975 (to D.A.H.). I.H.S. is supported by National Institutes of Health Pre-doctoral Fellowship NS063547, and by the Medical Scientist Training Program at Washington University.
References
- Prusiner SB. Prion Biology and Diseases 1999; Cold Spring Harbor Cold Spring Harbor Laboratory Press
- Prusiner SB. Prions. Proc Natl Acad Sci USA 1998; 95:13363 - 13383; PMID: 9811807; http://dx.doi.org/10.1073/pnas.95.23.13363
- Race R, Raines A, Raymond GJ, Caughey B, Chesebro B. Long-term subclinical carrier state precedes scrapie replication and adaptation in a resistant species: analogies to bovine spongiform encephalopathy and variant Creutzfeldt-Jakob disease in humans. J Virol 2001; 75:10106 - 10112; PMID: 11581378; http://dx.doi.org/10.1128/JVI.75.21.10106-12.2001
- Hill AF, Joiner S, Linehan J, Desbruslais M, Lantos PL, Collinge J. Species-barrier-independent prion replication in apparently resistant species. Proc Natl Acad Sci USA 2000; 97:10248 - 10253; PMID: 10963685; http://dx.doi.org/10.1073/pnas.97.18.10248
- Lasmézas CI, Deslys JP, Robain O, Jaegly A, Beringue V, Peyrin JM, et al. Transmission of the BSE agent to mice in the absence of detectable abnormal prion protein. Science 1997; 275:402 - 405; PMID: 8994041; http://dx.doi.org/10.1126/science.275.5298.402
- Manson JC, Jamieson E, Baybutt H, Tuzi NL, Barron R, McConnell I, et al. A single amino acid alteration (101L) introduced into murine PrP dramatically alters incubation time of transmissible spongiform encephalopathy. EMBO J 1999; 18:6855 - 6864; PMID: 10581259; http://dx.doi.org/10.1093/emboj/18.23.6855
- Büeler H, Fischer M, Lang Y, Bluethmann H, Lipp HP, DeArmond SJ, et al. Normal development and behaviour of mice lacking the neuronal cell-surface PrP protein. Nature 1992; 356:577 - 582; PMID: 1373228; http://dx.doi.org/10.1038/356577a0
- Harris DA, True HL. New insights into prion structure and toxicity. Neuron 2006; 50:353 - 357; PMID: 16675391; http://dx.doi.org/10.1016/j.neuron.2006.04.020
- Brandner S, Isenmann S, Raeber A, Fischer M, Sailer A, Kobayashi Y, et al. Normal host prion protein necessary for scrapie-induced neurotoxicity. Nature 1996; 379:339 - 343; PMID: 8552188; http://dx.doi.org/10.1038/379339a0
- Chesebro B, Race B, Meade-White K, Lacasse R, Race R, Klingeborn M, et al. Fatal transmissible amyloid encephalopathy: a new type of prion disease associated with lack of prion protein membrane anchoring. PLoS Pathog 2010; 6:1000800; PMID: 20221436; http://dx.doi.org/10.1371/journal.ppat.1000800
- Li A, Christensen HM, Stewart LR, Roth KA, Chiesa R, Harris DA. Neonatal lethality in transgenic mice expressing prion protein with a deletion of residues 105–125. EMBO J 2007; 26:548 - 558; PMID: 17245437; http://dx.doi.org/10.1038/sj.emboj.7601507
- Christensen HM, Harris DA. A deleted prion protein that is neurotoxic in vivo is localized normally in cultured cells. J Neurochem 2009; 108:44 - 56; PMID: 19046329; http://dx.doi.org/10.1111/j.1471-4159.2008.05719.x
- Massignan T, Stewart RS, Biasini E, Solomon IH, Bonetto V, Chiesa R, et al. A novel, drug-based, cellular assay for the activity of neurotoxic mutants of the prion protein. J Biol Chem 2010; 285:7752 - 7765; PMID: 19940127; http://dx.doi.org/10.1074/jbc.M109.064949
- Marcotti W, van Netten SM, Kros CJ. The aminoglycoside antibiotic dihydrostreptomycin rapidly enters mouse outer hair cells through the mechano-electrical transducer channels. J Physiol 2005; 567:505 - 521; PMID: 15994187; http://dx.doi.org/10.1113/jphysiol.2005.085951
- Myrdal SE, Steyger PS. TRPV1 regulators mediate gentamicin penetration of cultured kidney cells. Hear Res 2005; 204:170 - 182; PMID: 15925202; http://dx.doi.org/10.1016/j.heares.2005.02.005
- Massignan T, Biasini E, Harris DA. A Drug-Based Cellular Assay (DBCA) for studying cytotoxic and cytoprotective activities of the prion protein: A practical guide. Methods 2011; 53:214 - 219; PMID: 21115124; http://dx.doi.org/10.1016/j.ymeth.2010.11.005
- Solomon IH, Huettner JE, Harris DA. Neurotoxic mutants of the prion protein induce spontaneous ionic currents in cultured cells. J Biol Chem 2010; 285:26719 - 26726; PMID: 20573963; http://dx.doi.org/10.1074/jbc.M110.134619
- Solomon IH, Khatri N, Biasini E, Massignan T, Huettner JE, Harris DA. An N-terminal polybasic domain and cell surface localization are required for mutant prion protein toxicity. J Biol Chem 2011; 286:14724 - 14736; PMID: 21385869; http://dx.doi.org/10.1074/jbc.M110.214973
- Wadia JS, Schaller M, Williamson RA, Dowdy SF. Pathologic prion protein infects cells by lipid-raft dependent macropinocytosis. PLoS One 2008; 3:3314; PMID: 19390657; http://dx.doi.org/10.1371/journal.pone.0003314
- Gump JM, Dowdy SF. TAT transduction: the molecular mechanism and therapeutic prospects. Trends Mol Med 2007; 13:443 - 448; PMID: 17913584; http://dx.doi.org/10.1016/j.molmed.2007.08.002
- Warner RG, Hundt C, Weiss S, Turnbull JE. Identification of the heparan sulfate binding sites in the cellular prion protein. J Biol Chem 2002; 277:18421 - 18430; PMID: 11882649; http://dx.doi.org/10.1074/jbc.M110406200
- Taylor DR, Whitehouse IJ, Hooper NM. Glypican-1 mediates both prion protein lipid raft association and disease isoform formation. PLoS Pathog 2009; 5:1000666; PMID: 19936054; http://dx.doi.org/10.1371/journal.ppat.1000666
- Shyng SL, Heuser JE, Harris DA. A glycolipid-anchored prion protein is endocytosed via clathrin-coated pits. J Cell Biol 1994; 125:1239 - 1250; PMID: 7911471; http://dx.doi.org/10.1083/jcb.125.6.1239
- Shyng SL, Moulder KL, Lesko A, Harris DA. The N-terminal domain of a glycolipid-anchored prion protein is essential for its endocytosis via clathrin-coated pits. J Biol Chem 1995; 270:14793 - 14800; PMID: 7782345; http://dx.doi.org/10.1074/jbc.270.24.14793
- Harris DA. Trafficking, turnover and membrane topology of PrP. Br Med Bull 2003; 66:71 - 85; PMID: 14522850; http://dx.doi.org/10.1093/bmb/66.1.71
- Parkin ET, Watt NT, Turner AJ, Hooper NM. Dual mechanisms for shedding of the cellular prion protein. J Biol Chem 2004; 279:11170 - 11178; PMID: 14711812; http://dx.doi.org/10.1074/jbc.M312105200
- Shmerling D, Hegyi I, Fischer M, Blättler T, Brandner S, Götz J, et al. Expression of amino-terminally truncated PrP in the mouse leading to ataxia and specific cerebellar lesions. Cell 1998; 93:203 - 214; PMID: 9568713; http://dx.doi.org/10.1016/S0092-8674(00)81572-X
- Baumann F, Tolnay M, Brabeck C, Pahnke J, Kloz U, Niemann HH, et al. Lethal recessive myelin toxicity of prion protein lacking its central domain. EMBO J 2007; 26:538 - 547; PMID: 17245436; http://dx.doi.org/10.1038/sj.emboj.7601510
- Telling GC, Haga T, Torchia M, Tremblay P, DeArmond SJ, Prusiner SB. Interactions between wild-type and mutant prion proteins modulate neurodegeneration in transgenic mice. Genes Dev 1996; 10:1736 - 1750; PMID: 8698234; http://dx.doi.org/10.1101/gad.10.14.1736
- Rodriguez MM, Peoc'h K, Haïk S, Bouchet C, Vernengo L, Mañana G, et al. A novel mutation (G114V) in the prion protein gene in a family with inherited prion disease. Neurology 2005; 64:1455 - 1457; PMID: 15851745; http://dx.doi.org/10.1212/01.WNL.0000158618.39527.93
- Chiesa R, Piccardo P, Ghetti B, Harris DA. Neurological illness in transgenic mice expressing a prion protein with an insertional mutation. Neuron 1998; 21:1339 - 1351; PMID: 9883727; http://dx.doi.org/10.1016/S0896-6273(00)80653-4
- Dossena S, Imeri L, Mangieri M, Garofoli A, Ferrari L, Senatore A, et al. Mutant prion protein expression causes motor and memory deficits and abnormal sleep patterns in a transgenic mouse model. Neuron 2008; 60:598 - 609; PMID: 19038218; http://dx.doi.org/10.1016/j.neuron.2008.09.008
- Christensen HM, Dikranian K, Li A, Baysac KC, Walls KC, Olney JW, et al. A highly toxic cellular prion protein induces a novel, nonapoptotic form of neuronal death. Am J Pathol 2010; 176:2695 - 2706; PMID: 20472884; http://dx.doi.org/10.2353/ajpath.2010.091007
- Muramoto T, Scott M, Cohen FE, Prusiner SB. Recombinant scrapie-like prion protein of 106 amino acids is soluble. Proc Natl Acad Sci USA 1996; 93:15457 - 15462; PMID: 8986833; http://dx.doi.org/10.1073/pnas.93.26.15457
- Hölscher C, Delius H, Bürkle A. Overexpression of nonconvertible PrPC delta114–121 in scrapie-infected mouse neuroblastoma cells leads to trans-dominant inhibition of wild-type PrP(Sc) accumulation. J Virol 1998; 72:1153 - 1159; PMID: 9445012
- Norstrom EM, Mastrianni JA. The AGA AAA GA palindrome in PrP is required to generate a productive PrPSc-PrPC complex that leads to prion propagation. J Biol Chem 2005; 280:27236 - 27243; PMID: 15917252; http://dx.doi.org/10.1074/jbc.M413441200
- Trevitt CR, Collinge J. A systematic review of prion therapeutics in experimental models. Brain 2006; 129:2241 - 2265; PMID: 16816391; http://dx.doi.org/10.1093/brain/awl150
- Laurén J, Gimbel DA, Nygaard HB, Gilbert JW, Strittmatter SM. Cellular prion protein mediates impairment of synaptic plasticity by amyloid-beta oligomers. Nature 2009; 457:1128 - 1132; PMID: 19242475; http://dx.doi.org/10.1038/nature07761
- Resenberger UK, Harmeier A, Woerner AC, Goodman JL, Müller V, Krishnan R, et al. The cellular prion protein mediates neurotoxic signalling of β-sheet-rich conformers independent of prion replication. EMBO J 2011; 30:2057 - 2070; PMID: 21441896; http://dx.doi.org/10.1038/emboj.2011.86
- Yang W, Cook J, Rassbach B, Lemus A, DeArmond SJ, Mastrianni JA. A New Transgenic Mouse Model of Gerstmann-Straussler-Scheinker Syndrome Caused by the A117V Mutation of PRNP. J Neurosci 2009; 29:10072 - 10080; PMID: 19675240; http://dx.doi.org/10.1523/JNEUROSCI.2542-09.2009