Abstract
RNAs fold into intricate structures that are determined by specific base pairing interactions encoded within their primary sequences. Recently, a number of transcriptome-wide studies have suggested that RNA secondary structure is a potent cis-acting regulator of numerous post-transcriptional processes in viruses and eukaryotes. However, the need for experimentally-based structure determination methods has not been well addressed. Here, we show that the regulatory significance of Arabidopsis RNA secondary structure is revealed specifically through high-throughput, sequencing-based, structure mapping data, not by computational prediction. Additionally, we find that transcripts with similar levels of secondary structure in their UTRs (5' or 3') or CDS tend to encode proteins with coherent functions. Finally, we reveal that portions of mRNAs encoding predicted protein domains are significantly more structured than those specifying inter-domain regions. In total, our findings show the utility of high-throughput, sequencing-based, structure-mapping approaches and suggest that mRNA folding regulates protein maturation and function.
It is becoming increasingly clear that similar to proteins, RNAs must also fold into specific confirmations to function properly. This notion is emphasized by ribozymes and the known classes of structural RNAs (e.g., rRNAs, rRNAs; tRNAs, tRNAs; and small nuclear RNAs, snRNAs). For instance, rRNAs, tRNAs and snRNAs must adopt specific structural conformations to allow formation of functional ribosomes, enable amino acid addition during protein translation, and form competent spliceosomes, respectively.Citation1-Citation3 Similarly, long noncoding RNAs must adopt precise folding patterns to interact with regulatory proteins, which ultimately results in the modulation of expression from specific protein-coding loci.Citation4
Additionally, numerous single transcript studies indicate that structure is equally important in mRNA maturation and regulation.Citation5 For instance, secondary structure regulates the maturation of specific protein-coding mRNAs by effecting splicing and polyadenylation site accessibility.Citation6-Citation9 Moreover, RNA secondary structure regulates ribosome recruitment and the translation efficiency of specific transcripts by conferring additional requirements for initiation,Citation10,Citation11 or decreasing the rate of translation elongation.Citation12 Finally, various RNA structural elements have been found to regulate mRNA stability.Citation13 In total, these results have established that RNA folding can regulate nearly every point of the mRNA lifecycle.
Nevertheless, few studies have addressed the transcriptome-wide significance of structure in mRNA regulation and function. We sought to fill this gap by applying a high-throughput, sequencing-based, structure mapping approach to measure the folding patterns of every detectable transcript in unopened flower buds of Arabidopsis thaliana. Thus, we applied this technique in conjunction with sequencing of a number of different RNA populations (rRNA-depleted total RNA, RNA-seq; ribosome-bound RNA, ribo-seq; small RNA, smRNA-seq) to survey the functional relevance of structure across the Arabidopsis transcriptome.Citation14
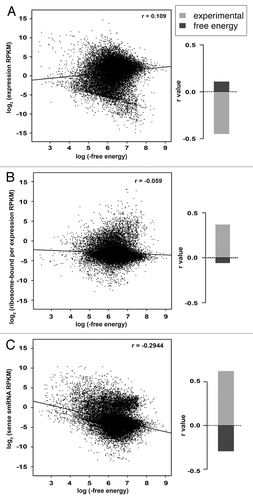
Our initial analysis revealed that RNA secondary structure has significant regulatory effects in Arabidopsis.Citation14 Thus, we wanted to determine if computationally predicted RNA secondary structure would also uncover similar regulatory significance for this feature. To test this, we calculated the free energy-based structure for all Arabidopsis transcripts using RNAFold.Citation15 We then determined the correlation between increasing computationally predicted RNA secondary structure and mRNA abundance, ribosome association and smRNA processing from these transcripts. We found a weak positive correlation (Pearson correlation r = 0.109) between computationally predicted structure values and overall mRNA abundance (). We also observed that these same structure values suggested a negative effect of RNA folding on mRNA association with ribosomes () and processing of these transcripts into smRNAs (). We then compared these findings to our results using experimentally-based structure mapping data.Citation14 We found that all of the correlations using computationally predicted structure are significantly weaker and of the opposite magnitude when compared with those from experimental data (, bar graphs). It is worth noting that the results for the computationally predicted RNA secondary structure also contradict previous qPCR-based validation in which five highly structured transcripts were significantly less abundant and more ribosome-bound than seven lowly structured transcripts.Citation14 In total, our results indicate that experimentally-based structure mapping data are necessary to uncover the regulatory functions of RNA folding in eukaryotic transcriptomes.
Figure 1. Experimentally determined structure data are better than free energy in predicting the regulatory significance of mRNA folding. For every detectable Arabidopsis transcript, RNAFold-predicted free energy (x-axis) is plotted against (A) average expression values as determined by RNA-seq (y-axis), (B) average ribosome occupancy as determined by normalizing ribo-seq to RNA-seq values (y-axis) and (C) average smRNA reads per transcript in the sense orientation (y-axis). To the right of each scatterplot is a comparison of r (Pearson correlation) values (y-axis) derived from these analyses using the RNAFold-predicted free energy (dark gray bar) or experimental structure dataCitation14 (light gray bar).
A previous gene ontology (GO) analysis suggested that transcripts with similar levels of overall experimentally determined secondary structure encoded proteins with coherent functions.Citation14 To expand and improve the resolution of these results, we sorted transcripts by total structure scores within specific mRNA regions [both UTRs (5′ and 3′) and CDS] and performed GO enrichment analysis on the 10% highest and 10% lowest structured transcripts for each segment. With this analysis, we also sought to reduce potential biases by reporting all GO terms at a specific level, as determined by a depth first search of the of the ontology tree. We observed enrichment of defense-related terms such as “cell killing,” “immune effector process” and “defense response to virus” among transcripts with high levels of structure in their CDS or 3′ UTR (), suggesting that structural elements in these regions are likely important regulators of the plant defense response.
Figure 2. Transcripts with similar levels of total structure in their UTRs (5′ or 3′) or CDS tend to encode proteins with coherent functions. Every detectable Arabidopsis transcript was sorted by mean structure in the 5′ UTR, CDS and 3′ UTR. The top 10% most structured and least structured transcripts were tested for enrichment of biological process gene ontology (GO) terms using the DAVID package.Citation21, Citation22 Output was filtered such that terms were retained only if they (1) were enriched at p-value < 0.05 for at least one structure category and (2) they were separated from the “biological process” ancestor term by no more than two parents (depth first search level ≤ 3). Each term was then hierarchically clustered based upon enrichment profiles and plotted as a heatmap (−log10[p-value]). No colored bar indicates that the enrichment is not significant.
![Figure 2. Transcripts with similar levels of total structure in their UTRs (5′ or 3′) or CDS tend to encode proteins with coherent functions. Every detectable Arabidopsis transcript was sorted by mean structure in the 5′ UTR, CDS and 3′ UTR. The top 10% most structured and least structured transcripts were tested for enrichment of biological process gene ontology (GO) terms using the DAVID package.Citation21, Citation22 Output was filtered such that terms were retained only if they (1) were enriched at p-value < 0.05 for at least one structure category and (2) they were separated from the “biological process” ancestor term by no more than two parents (depth first search level ≤ 3). Each term was then hierarchically clustered based upon enrichment profiles and plotted as a heatmap (−log10[p-value]). No colored bar indicates that the enrichment is not significant.](/cms/asset/c1d1f8e9-4c4e-4dd3-af56-74b49aca4c61/kpsb_a_10924301_f0002.gif)
Additionally, we observed enrichment of “response to inorganic substance” (), and corresponding enrichment of the child term “response to iron ion” (at level 5, data not shown) terms in transcripts with high levels of structure in their 5′ UTRs (). It is of note that this is consistent with known structural elements (iron-responsive elements or IREs) that are often found in the UTRs of transcripts involved in iron metabolism.Citation16 Interestingly, we also found enrichment for the term “gene silencing” () and the child term “RNA interference” (at level 5, data not shown) in transcripts with high structure in the 3′ UTR (). This result leads to the interesting speculation that transcripts involved in RNAi are themselves regulated through structural elements that are recognized by DICER-LIKE proteins resulting in smRNA production, which would ultimately form an autoregulatory loop.
Finally, “response to heat” was enriched in transcripts with highly structured 5′ UTRs (). One possible explanation for this observation is that structure may enable transcripts to directly sense heat. In support of this idea, bacterial transcripts involved in responding to heat stress unfold highly structured elements in their 5′ UTRs, which results in enhanced translation initiation from these mRNAs.Citation17 Future experiments will be needed to identify and determine the functional significance of the specific structural elements in these classes of highly folded transcripts.
Given our previous observations that lowly structured transcripts tend to be more abundant and degraded at a lower rate than highly structured transcripts,Citation14 we expected these mRNAs to encode proteins involved in constitutive processes. In support of this theory, transcripts with lowly structured UTRs were enriched for “photosynthesis” () and the child terms “photosynthetic electron transport chain” and “porphyrin metabolic process” (level 5, data not shown). Similarly, lowly structured transcripts are enriched for “generation of precursor metabolites and energy,” “gas transport,” and “pigment biosynthetic process” (). Nevertheless, we also observed terms that likely apply to inducible processes, such as “response to water,” “response to organic substance,” “response to osmotic stress,” and “response to cold” for transcripts with low levels of structure throughout their length (). Thus, low levels of secondary structure mark transcripts whose protein products are involved in both constitutive and inducible processes. In total, these results reveal that mRNAs encoding proteins with related functions tend to have common levels of RNA secondary structure in Arabidopsis. This is likely to provide a mechanism of regulation for mRNAs encoding proteins with coherent functions, as suggested by the post-transcriptional operon hypothesis.Citation18
Finally, we used our nucleotide-resolution structure dataCitation14 to test whether there is a correspondence between RNA and protein structure in Arabidopsis, as was previously observed for the HIV transcriptome.Citation19 This is pertinent because stable RNA secondary structure can reduce the rate of translation elongation, which may give nascent protein domains time to properly fold.Citation20 In support of this hypothesis, structure mapping of the HIV RNA genome revealed higher structure in regions encoding protein inter-domain linkers.Citation19 Thus, we sought to determine whether our Arabidopsis data follow a similar pattern. While few crystal structures exist for Arabidopsis proteins, domain structures can be predicted on the basis of homology to orthologs in the Protein Data Bank (PDB). We gathered protein domain annotation data sets from various prediction algorithms, and compared mean RNA secondary structure in domain-encoding and inter-domain regions within protein-coding mRNAs. Interestingly, we found that domain-encoding regions are significantly more structured across all prediction methods (). It is notable that we restricted our analysis specifically to the CDS of transcripts. Therefore, the structural difference we observed in domain-encoding mRNA regions cannot be attributed to higher overall structure in the CDS as compared with untranslated regions.Citation14 Furthermore, our results are consistent across multiple domain prediction algorithms, providing multiple observations of this finding. In total, these results reveal there is a signature of increased RNA secondary structure within domain-encoding portions of Arabidopsis mRNAs, which is opposite of what was observed for HIV.Citation19 These findings provide further support for the hypothesis that secondary structure is a significant feature for specifying foreign (e.g., viral pathogens) from self-encoded mRNAs in plant transcriptomes. Specifically, viruses seem to encode mRNAs with lower structure in domain-encoding portions,Citation19 while we have uncovered here that plant transcripts display the opposite pattern.
Figure 3. Regions of Arabidopsis mRNAs encoding predicted protein domains are significantly more structured than those specifying inter-domain portions. For every Arabidopsis transcript that is predicted to encode a protein domain (by numerous methods), the difference in mean RNA structure (y-axis) [as defined by (mean domain structure)-(mean non-domain structure)] is plotted. Protein domains were predicted from BLAST searches against various databases (x-axis, see www.ebi.ac.uk/Tools/pfa/iprscan/help/), and their annotations were downloaded from The Arabidopsis Information Resource (ftp://ftp.arabidopsis.org/home/tair/Proteins/Domains/OLD/TAIR9_all.domains). Grey dotted line denotes difference in mean RNA structure = 0 [(mean domain structure)-(mean non-domain structure)], gray dots indicate mean difference for each protein domain data set. * and *** denote p-value < 0.05 and < 10−10, respectively. p-values were calculated using a 2-tailed paired t-test.
![Figure 3. Regions of Arabidopsis mRNAs encoding predicted protein domains are significantly more structured than those specifying inter-domain portions. For every Arabidopsis transcript that is predicted to encode a protein domain (by numerous methods), the difference in mean RNA structure (y-axis) [as defined by (mean domain structure)-(mean non-domain structure)] is plotted. Protein domains were predicted from BLAST searches against various databases (x-axis, see www.ebi.ac.uk/Tools/pfa/iprscan/help/), and their annotations were downloaded from The Arabidopsis Information Resource (ftp://ftp.arabidopsis.org/home/tair/Proteins/Domains/OLD/TAIR9_all.domains). Grey dotted line denotes difference in mean RNA structure = 0 [(mean domain structure)-(mean non-domain structure)], gray dots indicate mean difference for each protein domain data set. * and *** denote p-value < 0.05 and < 10−10, respectively. p-values were calculated using a 2-tailed paired t-test.](/cms/asset/7e612f59-b324-46c5-8fd3-f5353c075268/kpsb_a_10924301_f0003.gif)
In conclusion, our results add to the growing body of evidence that RNA secondary structure is an important post-transcriptional regulatory element that affects all levels of gene expression. Importantly, we have revealed that RNA secondary structure marks groups of mRNAs encoding proteins with coherent functions (), as well as domain-encoding regions of transcripts (). Thus, it is likely that this feature also has broad effects on protein regulation and function in plants, which is a topic that needs to be further explored in the future.
| Abbreviations: | ||
| rRNA | = | ribosomal RNA |
| tRNA | = | transfer RNA |
| snRNA | = | small nuclear RNA |
| smRNA | = | small RNA |
| smRNA-seq | = | small RNA sequencing |
| GO | = | gene ontology |
| PDB | = | Protein Data Bank |
Acknowledgments
This work was funded by NSF Career Award MCB-1053846 to B.D.G., NHGRI 5T32HG000046-13 to F.L and NIGMS 5T32GM007229-37 to L.E.V. The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Related Research Data
References
- Buratti E, Baralle FE. Influence of RNA secondary structure on the pre-mRNA splicing process. Mol Cell Biol 2004; 24:10505 - 14; http://dx.doi.org/10.1128/MCB.24.24.10505-10514.2004; PMID: 15572659
- Schroeder R, Barta A, Semrad K. Strategies for RNA folding and assembly. Nat Rev Mol Cell Biol 2004; 5:908 - 19; http://dx.doi.org/10.1038/nrm1497; PMID: 15520810
- Trappl K, Polacek N. The ribosome: a molecular machine powered by RNA. Met Ions Life Sci 2011; 9:253 - 75; http://dx.doi.org/10.1039/9781849732512-00253; PMID: 22010275
- Khalil AM, Rinn JL. RNA-protein interactions in human health and disease. Semin Cell Dev Biol 2011; 22:359 - 65; http://dx.doi.org/10.1016/j.semcdb.2011.02.016; PMID: 21333748
- Silverman IM, Li F, Gregory BD. Genomic era analyses of RNA secondary structure and RNA-binding proteins reveal their significance to post-transcriptional regulation in plants. Plant Sci 2013; 205-206:55 - 62; http://dx.doi.org/10.1016/j.plantsci.2013.01.009; PMID: 23498863
- Raker VA, Mironov AA, Gelfand MS, Pervouchine DD. Modulation of alternative splicing by long-range RNA structures in Drosophila. Nucleic Acids Res 2009; 37:4533 - 44; http://dx.doi.org/10.1093/nar/gkp407; PMID: 19465384
- Warf MB, Berglund JA. Role of RNA structure in regulating pre-mRNA splicing. Trends Biochem Sci 2010; 35:169 - 78; http://dx.doi.org/10.1016/j.tibs.2009.10.004; PMID: 19959365
- Klasens BI, Das AT, Berkhout B. Inhibition of polyadenylation by stable RNA secondary structure. Nucleic Acids Res 1998; 26:1870 - 6; http://dx.doi.org/10.1093/nar/26.8.1870; PMID: 9518478
- Zarudnaya MI, Kolomiets IM, Potyahaylo AL, Hovorun DM. Downstream elements of mammalian pre-mRNA polyadenylation signals: primary, secondary and higher-order structures. Nucleic Acids Res 2003; 31:1375 - 86; http://dx.doi.org/10.1093/nar/gkg241; PMID: 12595544
- Kozak M. Leader length and secondary structure modulate mRNA function under conditions of stress. Mol Cell Biol 1988; 8:2737 - 44; PMID: 3405216
- Svitkin YV, Pause A, Haghighat A, Pyronnet S, Witherell G, Belsham GJ, et al. The requirement for eukaryotic initiation factor 4A (elF4A) in translation is in direct proportion to the degree of mRNA 5′ secondary structure. RNA 2001; 7:382 - 94; http://dx.doi.org/10.1017/S135583820100108X; PMID: 11333019
- Wen JD, Lancaster L, Hodges C, Zeri AC, Yoshimura SH, Noller HF, et al. Following translation by single ribosomes one codon at a time. Nature 2008; 452:598 - 603; http://dx.doi.org/10.1038/nature06716; PMID: 18327250
- Goodarzi H, Najafabadi HS, Oikonomou P, Greco TM, Fish L, Salavati R, et al. Systematic discovery of structural elements governing stability of mammalian messenger RNAs. Nature 2012; 485:264 - 8; http://dx.doi.org/10.1038/nature11013; PMID: 22495308
- Li F, Zheng Q, Vandivier LE, Willmann MR, Chen Y, Gregory BD. Regulatory impact of RNA secondary structure across the Arabidopsis transcriptome. Plant Cell 2012; 24:4346 - 59; http://dx.doi.org/10.1105/tpc.112.104232; PMID: 23150631
- Zuker M, Stiegler P. Optimal computer folding of large RNA sequences using thermodynamics and auxiliary information. Nucleic Acids Res 1981; 9:133 - 48; http://dx.doi.org/10.1093/nar/9.1.133; PMID: 6163133
- Leipuviene R, Theil EC. The family of iron responsive RNA structures regulated by changes in cellular iron and oxygen. Cell Mol Life Sci 2007; 64:2945 - 55; http://dx.doi.org/10.1007/s00018-007-7198-4; PMID: 17849083
- Lai EC. RNA sensors and riboswitches: self-regulating messages. Curr Biol 2003; 13:R285 - 91; http://dx.doi.org/10.1016/S0960-9822(03)00203-3; PMID: 12676109
- Tenenbaum SA, Christiansen J, Nielsen H. 2011. The Post-transcriptional Operon. In RNA, Vol. 703. Nielsen, H. & J. M. Walker, Eds.: 237-245. Humana Press.
- Watts JM, Dang KK, Gorelick RJ, Leonard CW, Bess JW Jr., Swanstrom R, et al. Architecture and secondary structure of an entire HIV-1 RNA genome. Nature 2009; 460:711 - 6; http://dx.doi.org/10.1038/nature08237; PMID: 19661910
- Kramer G, Boehringer D, Ban N, Bukau B. The ribosome as a platform for co-translational processing, folding and targeting of newly synthesized proteins. Nat Struct Mol Biol 2009; 16:589 - 97; http://dx.doi.org/10.1038/nsmb.1614; PMID: 19491936
- Huang W, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res 2009; 37:1 - 13; http://dx.doi.org/10.1093/nar/gkn923; PMID: 19033363
- Huang W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 2008; 4:44 - 57; http://dx.doi.org/10.1038/nprot.2008.211; PMID: 19131956