
Abstract
MATH-BTB proteins are known to act as substrate-specific adaptors of cullin3 (CUL3)-based ubiquitin E3 ligases to target protein for ubiquitination. In a previous study we reported the presence of 31 MATH-BTB genes in the maize genome and determined the regulatory role of the MATH-BTB protein MAB1 during meiosis to mitosis transition. In contrast to maize, there are only 6 homologous genes in the model plant Arabidopsis, while this family has largely expanded in grasses. Here, we report a phylogenetic analysis of the MATH-BTB gene family in 9 land plant species including various mosses, eudicots, and grasses. We extend a previous classification of the plant MATH-BTB family and additionally arrange the expanded group into 5 grass-specific clades. Synteny studies indicate that expansion occurred to a large extent due to local gene duplications. Expression studies of 3 closely related MATH-BTB genes in maize (MAB1–3) indicate highly specific expression pattern. In summary, this work provides a solid base for further studies comparing genetic and functional information of the MATH-BTB family especially in the grasses.
Proteins encompassing a MATH (Meprin and TRAF Homology) and a BTB (Bric-A-Brac, Tramtrack, Broad Complex) domain, also known as the POZ (Pox virus and Zinc finger) domain are commonly found in plant and animals, but not in fungi and serve in a wide range of cellular processes to regulate developmental processes and cell homeostasis.Citation1,Citation2 These 2 domains are found in many proteins of the MATH and BTB protein superfamilies, respectively. Each domain is capable to associate with a diverse set of other proteins. The vast majority MATH-BTB proteins contain an N-terminal MATH domain and a C-terminal BTB domain and are reported to function as substrate-specific adaptors in cullin3 (CUL3)-based E3 complexes (for a review see ref. Citation3).
Phylogenetic analyses of MATH-BTB genes in Arabidopsis and rice (Oryza sativa) revealed a large-scale expansion in the number of MATH-BTB encoding genes in the grass species. A total of 69 putative MATH-BTB genes were identified in rice, while Arabidopsis contains only 6 genes.Citation4,Citation5 Rice MATH-BTBs form 2 separate clades, a small core clade that is also associated with all 6 Arabidopsis MATH-BTBs and a large expanded group present only in rice.Citation5 Although the increase in number of MATH-BTB proteins implies a major significance of these proteins in rice, the functional linkage is not at all understood. Arabidopsis MATH-BTB proteins appear to possess a role in the regulation of transcription factors involved in ethylene and abscisic acid (ABA) signal transduction pathways and thus were discussed to play a role in abiotic stress responses.Citation6-Citation8
We previously reported the presence of 31 MATH-BTB genes in the maize (Zea mays) genome.Citation9 One of these proteins, MAB1, was analyzed in more detail and was shown to be specifically expressed in both germ lineages and the zygote, and has been shown to regulate spindle length during meiosis as well as nuclei identity during the first asymmetric pollen mitosis.Citation9 Since functional studies are missing for all other grass MATH-BTB proteins, a systematic classification of plant MATH-BTB proteins may contribute in elucidating functional redundancy among different MATH-BTB proteins. The availability of a large collection of genome sequences from various plant species prompted us to extend the first phylogenetic analysisCitation5 and to include additional eudicots, grass, and non-grass monocots as well as mosses as basal land plants.
For this study, we have first analyzed the genomes of 4 representatives of the grass family (Poaceae) for the presence of MATH-BTB genes: rice, Brachypodium distachyon, which is closely related to the Triticeae (wheat, barley), maize, and its close relative Sorghum bicolor. To further assess the evolutionary relationship of MATH-BTB genes in land plants (embryophytes) we expanded the analyses to the non-grass monocot banana (Musa acuminata), grapevine (Vitis vinfera) as another representative of the eudicot plants, and 2 representatives of basal land plants, namely a bryophyte (Physcomitrella patens) and a lycophyte (Selaginella moellendorffii). We performed exhaustive searches of several databases (e.g. Ensembl Plants, Phytozome, MSU Rice project) to identify MATH-BTB genes in these plant species. Our BLASTp and tBLASTn searches using known MATH-BTB sequences of Arabidopsis and maize as baits retrieved 115 newly identified MATH-BTB genes, of which 49 were identified in Brachypodium, 51 additional ones in Sorghum, 7 in banana, 5 in grapevine, and 3 additional ones in Selaginella (Table S1). These searches also recovered all previously identified sequences and resulted in a data set of 273 MATH-BTB genes in 9 plant genomes. About 90% of these genes belong to the 4 species of the grass family. The annotation of each MATH-BTB gene for each species and their assigned names can be found in Table S1. Additionally, few examples of genes encoding duplicated domains of either MATH or BTB domain, respectively, were found in each cereal genome. These are, for instance, GRMZM2G077951 containing a MATH-BTB-MATH-BTB structure, Sb06g029140, Sb01g021370, Sb01g021380, and Sb01g021410 displaying a MATH-BTB-BTB structure and Sb05g024820 with a MATH-MATH-BTB sequence. Additionally unusually long proteins of the MATH-BTB family were identified that do not contain any other recognizable domains except one MATH and one BTB domain (Os10g29495, BRADI5G09870, BRADI5G22350). These sequences were not included in phylogenetic analysis (see below) because their addition gave unstable results due to the effect of long-branch attraction.
Using the maximum likelihood method implemented in Seaview v.4.3.4Citation10 we generated a phylogentic tree supported with Shimodara-Hasegawa (SH)-test from 259 nucleotide sequences encoding full-length MATH-BTB proteins. As shown in all MATH-BTB genes from banana, Arabidopsis, grapevine, Selaginella, and Physcomitrella clustered into one clade, the core clade, together with 4 rice, 4 Sorghum, 4 Brachypodium, and 6 maize MATH-BTBs. The remaining 214 MATH-BTB genes entirely belonging to the grass family were distributed into 5 major subclades of the expanded clade (named as E1 to E5). While each expanded subclade was formed by less than 10 sequences from each grass species, subclades E4 and E5 in Sorghum and rice have further expanded (; Fig. S1). Particularly, 41 rice and 37 Sorghum genes dominated the E5 subclade comprised of 103 genes in the phylogram, while Sorghum also dominated the E4 subclade with 23 out of 49 genes. In contrast, only 3 maize MATH-BTB sequences clustered into the E4 subclade and none was found in subclade E3. In comparison to the core clade, the expanded clade showed significant expansion for each cereal species. Gingerich et al.Citation5 suggested that the expanded group is monocot specific. To further analyze this hypothesis, we included banana MATH-BTB genes from a non-grass monocot in the phylogram. We found that all banana MATH-BTB genes belong to the core clade further supporting the hypothesis that the expanded clade is grass-specific. The finding that 3 rounds of whole-genome duplication in the Musa lineage,Citation11 occurring independently of the Poales lineage, did not increase the number of MATH-BTB genes further strengthens the idea that the evolution and expansion of the MATH-BTB family in the grasses maybe attributed at least partly with grass-specific functions.
Figure 1. Unrooted phylogentic tree of 259 genes encoding MATH-BTB proteins from 9 representative land plant species (embryophyta). Analysis was done with Seaview v.4.3.4. using nucleotide sequence regions (open reading frames) of full-length MATH-BTB proteins with both MATH and BTB domains. Genes are color-coded by species (see legend for colors at the bottom right side of the tree). E1 to E5 indicate 5 major grass-specific subclades of the expanded clade. Core clade MATH-BTB genes form a separate clade. The number after decimal point for designated gene presents a splicing variant used for phylogenetic analysis. The bar of 0.5 is a branch length, which represents nucleotide substitutions per site. For sequence identifiers see Table S1. For statistical support of phylogenetic tree see Figure S1.
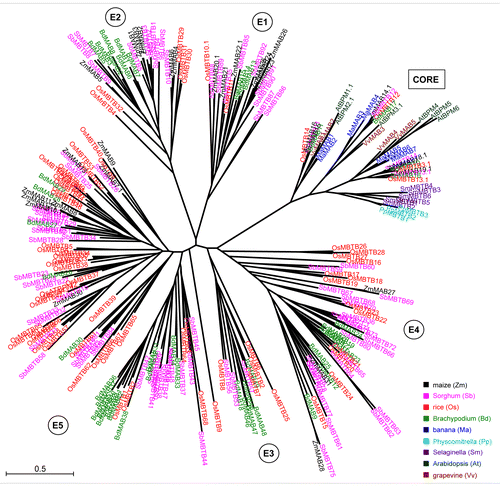
The number of Sorghum genes is almost 3 times higher than the number of maize genes, suggesting that the size of the maize MATH-BTB family had significantly reduced after the divergence of the 2 C4 plants Sorghum and maize. A likely scenario could be associated with the recent multiproliferation of retrotransposable elements especially in the maize lineage, which occurred within the last 1 million of yearsCitation12 and might have been associated with extensive gene losses after recombination events. Alternatively, a significant expansion of Sorghum and rice genes in E4 and E5 subclades, respectively, might have occurred due to yet unknown specific functions in these lineages. Besides 69 MATH-BTB genes, 5 MATH-related-BTB genes and 41 pseudogenes were reported as part of the MATH-BTB family in rice.Citation5 We assume a similar situation in the Sorghum genome. In database searches, we found 92 predicted protein-coding genes containing both MATH and BTB domains, respectively. However, since genomes are annotated by gene finder programs, which typically do not predict pseudogenes,Citation13 a separate effort should be attributed to manually curate pseudogenes. We noticed truncated ORFs for some of the Sorghum MATH-BTB genes or differing numbers of exons between duplicated genes in tandem duplicated regions, which are indications of potential pseudogenes. However, in order to avoid over predicting pseudogenes and discarding genuinely functional genes, we included all identified genes in our analysis. Notably more than half of rice MATH-BTB pseudogenes are transcribed.Citation5 This could have significant implications taken into account an interesting hypothesis of Sasidharan and GersteinCitation14 proposing that expressed pseudogenes can be processed into short interfering RNAs to regulate homologous coding genes. The maize genome, for example, contains a lower number of MATH-BTB genes compared with the other 3 cereals analyzed in this study, but overall maize has a higher number of splicing variants. Thus, it is tempting to speculate that maize substituted a possible gene loss by increasing the number of splicing variants, which can exert regulatory effects on full-length variants. This mode of regulation seems to be especially prominent for the only 2 human MATH-BTB genes, with one of them generating up to 23 splicing variants according to the Ensembl database.
MATH-BTB genes of the core clade are conserved in all 9 land plant species analyzed, indicating that loss of these genes might be associated with a fitness and/or viability disadvantage. This hypothesis is supported by the finding that protein sequences within this clade from different plant species shareCitation3 60 to 84% amino acid identity with almost no gaps in a sequence alignment. In contrast the grass-specific expanded clade shows more extended gaps and its members are less homologous to each other (on averageCitation3 45% identical at the amino acid level calculated by BLASTp multiple alignment tool). Our results thus corroborate the previous finding that the core group genes show highest degree of evolutionary conservation.Citation5 It was also hypothesized that conserved clades containing genes from several species display significant functional conservation and possibly regulate ancient pathways in plant development or physiology.Citation15,Citation16 So far, the only core genes functionally analyzed are from Arabidopsis with a role in the transcriptional regulation of abscisic acid signaling and fatty acid metabolism.Citation7,Citation8 Therefore, other candidate genes of the core group have to be analyzed at the functional and mechanistical level to sustain this hypothesis. It will also be interesting now to elucidate how general functions such as the regulation of spindle length during meiosis and nuclei identity during the first asymmetric pollen mitosis, which is achieved by the highly regulated MATH-BTB gene MAB1 of the expanded clade of maizeCitation9 is regulated in species such as Arabidopsis, which contains only 6 MATH-BTB genes displaying a very broad expression pattern.Citation17
A phylogeny based sequence alignment study alone cannot answer the question how the MATH-BTB family evolved and expanded in the grasses. To explore the evolutionary history of the expanded group, we searched for the occurrence of conserved synteny by using default parameters of BLASTp queries in Phytozome (www.phytozome.net), a comparative database for plant genomics. The matching groups with the smallest E-value are shown in . We complemented the results with the analysis of chromosome location of MATH-BTB genes (see also Table S1). The majority (around 80%) of Sorghum, Brachypodium, and rice MATH-BTB genes of the extended clade are located in gene clusters or are located on the same chromosome in close proximity. This finding indicates that the origin of the clade-specific expansion of MATH-BTB genes occurred to a large extend due to tandem or segmental duplication events. Additionally, with the exception of Sorghum many of the expanded genes are located in regions with a high content of transposable elements, which is particularly evident for maize. Only a minor number of expanded clade genes are present as isolated single copy genes. In contrast, all core MATH-BTB genes are single copies scattered to different chromosomal locations (Table S1).
Figure 2. Graphic display of shared synteny and tandem duplications of selected grass MATH-BTB genes from the expanded subclades E1-E5 of rice, Brachypodium, Sorghum, and maize. MATH-BTB genes are depicted with the label MB, BTB genes with B, MATH domain encoded genes with M, and known MATH-BTB pseudogenes in rice with P. These genes are additionally highlighted in red. Selected members of the MATH-BTB family are colored in gray and appear in the center of the diagram surrounded by 5 upstream and downstream neighbor genes. Note that some MATH-BTB genes identified in this study are depicted in other colors. The graph was generated using the Phytozome database. To simplify visualization of genes from different species belonging to the same family other than MATH-BTBs, they were labeled with numbers and appear in the same color. For corresponding sequence identifiers matching labels see Table S2.

Regarding synteny, we found that at least one gene from each species in extended subclades E1-E5 is located in microsyntenic chromosomal regions (). This finding suggests that the expanded group of MATH-BTB family was already formed in the common ancestor of cereals by at least 5 genes on different chromosome locations. However, we also observed that even though the genomic neighborhood of the representative MATH-BTB genes is preserved, gene order and orientation is not. This could be a consequence of rapid divergence of each subgroup and their variations due to species-specific gene loss and gain. Additionally, both maize and Brachypodium genomes underwent large-scale rearrangements and chromosomal reshuffling events that eventually led to a reduction in chromosome number.Citation18,Citation19 The complex evolution of the maize genome, involving allotetraploidization followed by extensive gene losses, subsequent chromosome breakage and fusion and numerous transpositions of retroelements, makes it difficult to align its chromosomal segments and to perform a comprehensive analysis possible in other grass species. Contrary, rice and Sorghum genomes have preserved much of the common ancestral genome structure of the grasses.Citation20 For that reason, when interpreting future results of shared synteny priority should be given to rice and Sorghum, and to a lesser extent to maize.
In this study we have further determined that the only functionally analyzed gene of the expanded clade, MAB1 of maize,Citation9 belongs to the E2 subclade (; Fig. S1). This subclade was therefore analyzed in more detail. Genes of the rice and Sorghum E2 subclade were found to occur in gene clusters in a tandem array with, for example, 10 genes in a subtelomeric region of chromosome 7 in Sorghum and 5 genes, 3 pseudogenes and a gene encoding only a BTB domain protein at a similar region of chromosome 8 in rice (Fig. S2). It is thus very likely that all members of the E2 group derived from an ancestral tandem duplication of a single gene locus. Rice chromosome 8 and Sorghum chromosome 7 represent homeologous chromosomes and share synteny with Brachypodium chromosome 3 and maize chromosomes 1 and 4. E2 subclade genes of maize and Brachypodium are scattered on 3 locations. Gene clusters of maize E2 subclade members on chromosome 1 and 4 can be explained by the fact that most chromosomal regions in rice correspond to 2 regions in maize as a result of its allopolyploidy origin. Thus, the chromosome location revealed that MAB1 closest homologs MAB2 and MAB3 are part of this ancestral syntenic region, while the location of MAB1 gene on chromosome 7 appears to originate from a (retro)transposition. Similarly to MAB1, one Brachypodium gene (BdMAB1) is located as a single copy on chromosome 2 with no shared synteny, implying that this gene might also represent a transposed duplicated gene. Other Brachypodium genes are located on 2 neighboring clusters on chromosome 3. It seems that these separated clusters have evolved by segmental insertion into a single cluster during Brachypodium genome rearrangements.
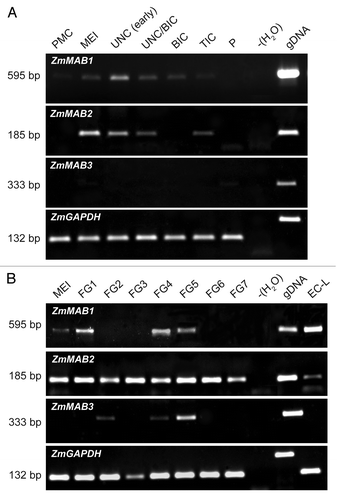
To analyze whether the most closely related maize genes MAB1, MAB2, and MAB3 share a similar expression pattern and may be able to complement each other, we first studied their expression pattern using various reproductive tissues and leaves as a vegetative tissue. Total RNA was extracted from defined stages of maize ovules and anthers as described in Juranić et al.Citation9 Primer sequences can be found in Supplemental Methods. As shown in similarly to MAB1, MAB2, and MAB3 are apparently germline-specific genes, and lack expression in vegetative tissues such as leaves (data not shown). However, they display significant different expression profiles indicating strictly regulated temporal expression and likely different functions. MAB1 is most strongly expressed during male germline development at the early uni-nucleate stage, MAB2 is expressed most strongly during male meiosis, while MAB3 expression is almost undetectable with faint signals at pollen maturity. During female germline development, MAB1 is again most strongly expressed after completing meiosis and in egg cells, while MAB2 shows a rather constitutive expression pattern and MAB3 is most abundant at stage FG5 when cellularization takes place. Moreover, MAB3 transcripts appear absent in egg cells. Synteny analysis and the relatively broad expression pattern of MAB2 suggest that it is most likely the parental gene of MAB1, which after a duplication event underwent neo- and/or subfunctionalization. According to the results using the BLASTp multiple alignment tool, MAB2 and MAB3 share 84% amino acid sequence identity among each other and 70% similarity with MAB1. The low expression of MAB3 could be explained through functional redundancy partly overlapping with MAB1 and MAB2.
Figure 3. Expression of ZmMAB1–3 during germline developmental in maize inbred line A188. (A) Expression during male gametophyte development from pollen mother cell (PMC), meiosis (MEI), uninucleate microspore (UNC), bicellular pollen (BIC), tricellular pollen (TIC), and mature pollen at shedding phase (P; anthesis). (B) Expression during female gametophyte development from meiosis (MEI) to mature female gametophytes (stage FG7). Stages after Evans and Grossniklaus.Citation21 An egg cell library (EC-L;Citation22) was included as a positive control for ZmMAB1. RT-PCR with water (-H20) served as negative, and genomic DNA (gDNA) as a positive control for all genes. Maize GAPDH was used as a housekeeping control.
The known function of MAB1 in the regulation of meiotic and mitotic divisions during germline development and a possible role during early embryogenesisCitation9 together with the finding that its closest paralogues genes MAB2 and MAB3 also display a germline-specific expression pattern as well as a probable single locus origin raises the intriguing possibility that the whole E2 subclade might be expressed in the germlines with conserved functions in reproduction. It is therefore important now to study the functions of many more grass-specific MATH-BTB genes from different subclades of the expanded as well as of the core clade. It will also be exciting to find out whether plant species containing only core clade MATH-BTB genes use these proteins to regulate the same conserved reproductive processes such as spindle length during meiosis to mitosis transition, which are regulated by expanded clade members in the grasses. In conclusion, we have presented here a tool and solid basis for more elaborate studies on the evolutionary pattern of grass MATH-BTB genes and for future functional studies in different land plant species.
| Abbreviations: | ||
| BTB | = | Bric-A-Brac, Tramtrack, Broad Complex protein domain |
| E1-E5 | = | MATH-BTB subclades of the expanded clade |
| MATH | = | Meprin and TRAF Homology protein domain |
Additional material
Download Zip (620.7 KB)Acknowledgments
Nadia G Krohn is acknowledged for providing cDNA samples of female gametophyte stages. We are grateful for an Alexander von Humboldt-Foundation institute partnership and the Ministry of Science, Education, and Sports of the Republic of Croatia (Project 119–1191196–1225) to support the exchange of knowledge and scientists between both labs.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Materials
Supplemental materials may be found here: www.landesbioscience.com/journals/psb/article/28242
References
- Stogios PJ, Downs GS, Jauhal JJS, Nandra SK, Privé GG. Sequence and structural analysis of BTB domain proteins. Genome Biol 2005; 6:R82; http://dx.doi.org/10.1186/gb-2005-6-10-r82; PMID: 16207353
- Zapata JM, Martínez-García V, Lefebvre S. Phylogeny of the TRAF/MATH domain. In: TNF receptor associated factors (TRAFs). Wu H, ed. New York: Springer, 2007; 597:1-24.
- Genschik P, Sumara I, Lechner E. The emerging family of CULLIN3-RING ubiquitin ligases (CRL3s): cellular functions and disease implications. EMBO J 2013; 32:2307 - 20; http://dx.doi.org/10.1038/emboj.2013.173; PMID: 23912815
- Gingerich DJ, Gagne JM, Salter DW, Hellmann H, Estelle M, Ma L, Vierstra RD. Cullins 3a and 3b assemble with members of the broad complex/tramtrack/bric-a-brac (BTB) protein family to form essential ubiquitin-protein ligases (E3s) in Arabidopsis.. J Biol Chem 2005; 280:18810 - 21; http://dx.doi.org/10.1074/jbc.M413247200; PMID: 15749712
- Gingerich DJ, Hanada K, Shiu SH, Vierstra RD. Large-scale, lineage-specific expansion of a bric-a-brac/tramtrack/broad complex ubiquitin-ligase gene family in rice. Plant Cell 2007; 19:2329 - 48; http://dx.doi.org/10.1105/tpc.107.051300; PMID: 17720868
- Weber H, Hellmann H. Arabidopsis thaliana BTB/ POZ-MATH proteins interact with members of the ERF/AP2 transcription factor family. FEBS J 2009; 276:6624 - 35; http://dx.doi.org/10.1111/j.1742-4658.2009.07373.x; PMID: 19843165
- Lechner E, Leonhardt N, Eisler H, Parmentier Y, Alioua M, Jacquet H, Leung J, Genschik P. MATH/BTB CRL3 receptors target the homeodomain-leucine zipper ATHB6 to modulate abscisic acid signaling. Dev Cell 2011; 21:1116 - 28; http://dx.doi.org/10.1016/j.devcel.2011.10.018; PMID: 22172674
- Chen L, Lee JH, Weber H, Tohge T, Witt S, Roje S, Fernie AR, Hellmann H. Arabidopsis BPM proteins function as substrate adaptors to a cullin3-based E3 ligase to affect fatty acid metabolism in plants. Plant Cell 2013; 25:2253 - 64; http://dx.doi.org/10.1105/tpc.112.107292; PMID: 23792371
- Juranič M, Srilunchang KO, Krohn NG, Leljak-Levanic D, Sprunck S, Dresselhaus T. Germline-specific MATH-BTB substrate adaptor MAB1 regulates spindle length and nuclei identity in maize. Plant Cell 2012; 24:4974 - 91; http://dx.doi.org/10.1105/tpc.112.107169; PMID: 23250449
- Gouy M, Guindon S, Gascuel O. SeaView version 4: A multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol Biol Evol 2010; 27:221 - 4; http://dx.doi.org/10.1093/molbev/msp259; PMID: 19854763
- D’Hont A, Denoeud F, Aury JM, Baurens FC, Carreel F, Garsmeur O, Noel B, Bocs S, Droc G, Rouard M, et al. The banana (Musa acuminata) genome and the evolution of monocotyledonous plants. Nature 2012; 488:213 - 7; http://dx.doi.org/10.1038/nature11241; PMID: 22801500
- Du C, Swigonová Z, Messing J. Retrotranspositions in orthologous regions of closely related grass species. BMC Evol Biol 2006; 6:62; http://dx.doi.org/10.1186/1471-2148-6-62; PMID: 16914031
- Thibaud-Nissen F, Ouyang S, Buell CR. Identification and characterization of pseudogenes in the rice gene complement. BMC Genomics 2009; 10:317; http://dx.doi.org/10.1186/1471-2164-10-317; PMID: 19607679
- Sasidharan R, Gerstein M. Genomics: protein fossils live on as RNA. Nature 2008; 453:729 - 31; http://dx.doi.org/10.1038/453729a; PMID: 18528383
- Thomas JH. Adaptive evolution in two large families of ubiquitin-ligase adapters in nematodes and plants. Genome Res 2006; 16:1017 - 30; http://dx.doi.org/10.1101/gr.5089806; PMID: 16825662
- Navarro-Quezada A, Schumann N, Quint M. Plant F-box protein evolution is determined by lineage-specific timing of major gene family expansion waves. PLoS One 2013; 8:e68672; http://dx.doi.org/10.1371/journal.pone.0068672; PMID: 23904908
- Weber H, Bernhardt A, Dieterle M, Hano P, Mutlu A, Estelle M, Genschik P, Hellmann H. Arabidopsis AtCUL3a and AtCUL3b form complexes with members of the BTB/POZ-MATH protein family. Plant Physiol 2005; 137:83 - 93; http://dx.doi.org/10.1104/pp.104.052654; PMID: 15618422
- Wei F, Coe E, Nelson W, Bharti AK, Engler F, Butler E, Kim H, Goicoechea JL, Chen M, Lee S, et al. Physical and genetic structure of the maize genome reflects its complex evolutionary history. PLoS Genet 2007; 3:e123; http://dx.doi.org/10.1371/journal.pgen.0030123; PMID: 17658954
- International Brachypodium Initiative. Genome sequencing and analysis of the model grass Brachypodium distachyon. Nature 2010; 463:763 - 8; http://dx.doi.org/10.1038/nature08747; PMID: 20148030
- Salse J, Abrouk M, Murat F, Quraishi UM, Feuillet C. Improved criteria and comparative genomics tool provide new insights into grass paleogenomics. Brief Bioinform 2009; 10:619 - 30; http://dx.doi.org/10.1093/bib/bbp037; PMID: 19720678
- Evans MMS, Grossniklaus U. (2009). The maize megagametophyte. In: Handbook of maize: its biology. Bennetzen JL and Hake SC. New York: Springer, 79-104.
- Dresselhaus T, Lörz H, Kranz E. Representative cDNA libraries from few plant cells. Plant J 1994; 5:605 - 10; http://dx.doi.org/10.1046/j.1365-313X.1994.5040605.x; PMID: 8012409