Abstract
Fanconi anemia (FA) is a heterogeneous disease associated with a bone marrow failure, cancer predisposition and hypersensitivity to DNA crosslinking agents. To date, 15 different genes have been shown to cause FA, all of which have some role in repair of defective DNA interstrand crosslinks. On a biochemical level, many FA individuals display insufficient growth hormone production, abnormal glucose or insulin metabolism. Clinical phenotype may include hydrocephalia, the erythrophagocytosis and diabetes mellitus, thus linking FA with metabolic disorders that involve impaired oxygen metabolism and mitochondrial alterations. Our recent study demonstrates the decrease of FA mitochondrial membrane potential, low ATP production, impaired oxygen uptake and pathological changes in the morphology of FA mitochondria. This is accompanied by inactivation of the enzymes responsible for energy production and detoxification of ROS. We also propose that FA oversensitivity to DNA crosslinkers may be caused by the overproduction of mitochondrial ROS.
Fanconi anemia (FA) is a rare autosomal recessive genetic disorder associated with a bone marrow failure, cancer predisposition and hypersensitivity to DNA crosslinking agents. Fifteen FA complementation groups have been characterized so far (A, B, C, D1/BRCA2, D2, E, F, G, I, J/BACH1/BRIP1, L, M, N/PALB2, O/RAD51C and FANCP/SLX4).Citation1 According to current model, the central regulatory event in the FA pathway is assembling FA proteins from subgroups A-, -B, -C, -E, -F, -G, -L and -M (FA core complex) in response to replicative stress followed by phosphorylation/monoubiquitylation of FANCD2–FANCI and recruitment of specific nucleases and polymerases required for the DNA repair.Citation2 Such stress can be caused by cross-linking agents and/or ROS. Interestingly, a number of FA patients reveal hydrocephalia, ventriculoperitoneal shunts, the erythrophagocytosis and type II diabetes mellitus—common hallmarks of diseases that involve impaired oxygen metabolisms and mitochondrial damage.Citation3 Initially adduced by NordensonCitation4 and by Joenje et al.,Citation5,Citation6 recent works confirmed an increased ROS in FA,Citation7 interference with cellular redox state and ATP production,Citation8 sensitivity to oxidant stimuli,Citation9,Citation10 accumulation of oxidized proteinsCitation11 and oxidative DNA damage.Citation12-Citation14 All these facts may suggest engagement of mitochondria to cope with increased ROS production in FA. Indeed, Mukhopadhyay et al.Citation15 found that the FANCG protein is localized in mitochondria and interacts with the mitochondrial peroxidase peroxyredoxin 3 (PRDX3). In turn, cells from the FA-A and FA-C subtypes also had PRDX3 cleavage and decreased peroxidase activity. These findings further supported the idea of mitochondria involvement in the pathogenesis of FA.
Our recently published study demonstrates that FA cells from at least groups A, C and D2 have not only high ROS level and low ΔѰm, but also reveal altered mitochondrial morphology accompanied by decreased ATP production and oxygen uptake.Citation16 Unlike physiologically normal conditions in which mitochondria are active in ATP synthesis and most of the oxygen consumed by the respiratory chain is completely reduced to water, accumulation of stalled replication forks in FA cells attracts various DNA repair enzymes leading to overproduction of ROS, interfering with cellular antioxidant defense mechanisms and resulting in chronic damage of the major mitochondrial functions: ATP syntheses, ROS production/detoxification and maintenance of ΔѰm and OCR. Indeed, we found impaired response to ROS of major mitochondrial complexes, especially detoxifying enzyme superoxide dismutase SOD1. If overexpressed, however, this could rescue oxygen uptake. Thus it seems likely that FA proteins and associated partners also act outside of the canonical Fanconi pathway, in particular, helping to neutralize ROS. Our research on mitochondrial fractionation experiments coupled with 2D-DIGE based technique will likely help to elucidate mitochondrial components that are affected in FA. Importantly, animal studies are required to recapitulate our in vitro findings with in vivo models.
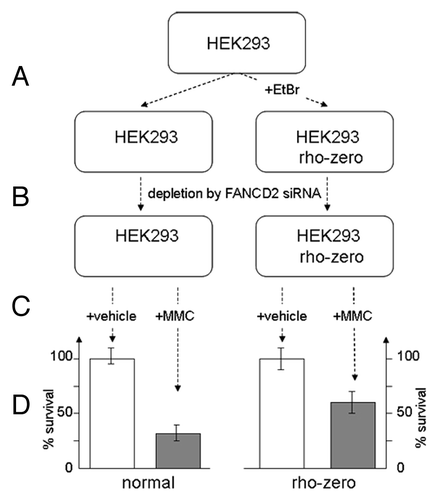
The concept of FA cells oversensitivity to cross-linking agents like MMC relies on inability of FA DNA repair machinery to overcome ICL-induced stalled replication blocks. In our study, we demonstrate that sensitivity of FA cells to crosslinker MMC can be reduced in the presence of ROS scavenger NAC suggesting that ROS evoked directly from MMC may equally contribute to MMC-induced cytotoxicity. Similar results can be obtained in MMC-sensitivity assay of human fibroblasts depleted for FANCD2 or FANCG by corresponding siRNAs. Cells pretreated with NAC display higher survival rate upon MMC treatment. Yet, the important question is whether mitochondria are involved in these processes. Since mitochondria are not only the first source of ROS formation but also the most susceptible ROS target, accumulation of MMC-induced ROS may further disable proper function of mitochondria by inactivating antioxidant defense mechanisms. In turn, removal the ROS associated with dysfunctional mitochondria may increase cell survival. In order to test this hypothesis, we prepared population of HEK293 cells with non-functional mitochondria by incubating them with a low concentration of ethidium bromide (). Such rho-zero cells displayed low sensitivity to MMC, as in case of normal cells. However, when converted to FA-like phenotype by corresponding treatment with FANCD2 siRNA (), rho-zero cells revealed almost two times less response to MMC vs. cells with fully functional mitochondria (). These experiments suggest that FA mitochondria can impact overall sensitivity of FA cells to MMC-induced ROS.
Figure 1. FA-like cells with non-functional mitochondria are less sensitive to MMC. (A) Rho-zero cells were prepared by incubating HEK 293 cells with ethidium bromide. (B) Depletion of cells from FANCD2 has been performed by double transfection with FANCD2 siRNA. (C) Cells were incubated for 1 h with 0.5uM MMC (final) or vehicle and MMC sensitivity assay was performed following 24 h post-treatment. (D) Cell sensitivity to MMC is increased for mitochondria-depleted rho-zero HEK293 cells vs. cells with functional mitochondria.
On a practical side, this may be amenable for patients requiring bone marrow transplantation and undergoing conditioning regimens that involve chemotherapy with cross-linking drugs. High sensitivity of FA cells to DNA damaging drugs can be decreased by combinatorial treatment with proper ROS scavengers. Furthermore, oxidative stress from accumulated ROS has been associated with tumor formation hence ROS-quenching compounds may delay tumor onset in FA. This can be especially relevant to FA cancer patients. Higher ROS production increases cancer cell dependence on ROS scavenging systems. Several studies suggested that therapies aimed at reducing ROS coming from extra mitochondrial source might offer effective means of combating malignancies. Essential to developing these therapeutic strategies is to maintain physiologically low ROS levels in normal tissues while inducing ROS in cancer cells. In this respect, screening a chemical library of small molecules—artificial analogs of a second antioxidant system (tocopherol, ascorbic acid, glutathione, bilirubin, etc.) or natural compounds that reduce oxidative DNA damage may hold promise for novel FA therapies.
| Abbreviations: | ||
| FA | = | Fanconi anemia |
| ROS | = | reactive oxygen species |
| ΔѰm | = | mitochondrial transmembrane potential |
| ICL | = | interstrand cross-links |
| 2D-DIGE | = | two-dimensional differential in gel electrophoreses, MMC, mitomycin C |
| NAC | = | N-Acetyl-l-cysteine |
| HEK | = | human embryonic kidney |
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- D’Andrea AD. Susceptibility pathways in Fanconi’s anemia and breast cancer. N Engl J Med 2010; 362:1909 - 19; http://dx.doi.org/10.1056/NEJMra0809889; PMID: 20484397
- Deans AJ, West SC. DNA interstrand crosslink repair and cancer. Nat Rev Cancer 2011; 11:467 - 80; http://dx.doi.org/10.1038/nrc3088; PMID: 21701511
- Pagano G, Talamanca AA, Castello G, Pallardó FV, Zatterale A, Degan P. Oxidative stress in Fanconi anaemia: from cells and molecules towards prospects in clinical management. Biol Chem 2012; 393:11 - 21; http://dx.doi.org/10.1515/BC-2011-227; PMID: 22628295
- Nordenson I. Effect of superoxide dismutase and catalase on spontaneously occurring chromosome breaks in patients with Fanconi’s anemia. Hereditas 1977; 86:147 - 50; http://dx.doi.org/10.1111/j.1601-5223.1977.tb01223.x; PMID: 914646
- Joenje H, Arwert F, Eriksson AW, de Koning H, Oostra AB. Oxygen-dependence of chromosomal aberrations in Fanconi’s anaemia. Nature 1981; 290:142 - 3; http://dx.doi.org/10.1038/290142a0; PMID: 7207594
- Dallapiccola B, Porfirio B, Mokini V, Alimena G, Isacchi G, Gandini E. Effect of oxidants and antioxidants on chromosomal breakage in Fanconi anemia lymphocytes. Hum Genet 1985; 69:62 - 5; http://dx.doi.org/10.1007/BF00295530; PMID: 3967890
- Degan P, Bonassi S, De Caterina M, Korkina LG, Pinto L, Scopacasa F, et al. In vivo accumulation of 8-hydroxy-2′-deoxyguanosine in DNA correlates with release of reactive oxygen species in Fanconi’s anaemia families. Carcinogenesis 1995; 16:735 - 41; http://dx.doi.org/10.1093/carcin/16.4.735; PMID: 7728950
- Bogliolo M, Borghini S, Abbondandolo A, Degan P. Alternative metabolic pathways for energy supply and resistance to apoptosis in Fanconi anaemia. Mutagenesis 2002; 17:25 - 30; http://dx.doi.org/10.1093/mutage/17.1.25; PMID: 11752230
- Rani R, Li J, Pang Q. Differential p53 engagement in response to oxidative and oncogenic stresses in Fanconi anemia mice. Cancer Res 2008; 68:9693 - 702; http://dx.doi.org/10.1158/0008-5472.CAN-08-1790; PMID: 19047147
- Saadatzadeh MR, Bijangi-Vishehsaraei K, Hong P, Bergmann H, Haneline LS. Oxidant hypersensitivity of Fanconi anemia type C-deficient cells is dependent on a redox-regulated apoptotic pathway. J Biol Chem 2004; 279:16805 - 12; http://dx.doi.org/10.1074/jbc.M313721200; PMID: 14764578
- Lyakhovich A, Surrallés J. Constitutive activation of caspase-3 and Poly ADP ribose polymerase cleavage in fanconi anemia cells. Mol Cancer Res 2010; 8:46 - 56; http://dx.doi.org/10.1158/1541-7786.MCR-09-0373; PMID: 20068062
- Du W, Rani R, Sipple J, Schick J, Myers KC, Mehta P, et al. The FA pathway counteracts oxidative stress through selective protection of antioxidant defense gene promoters. Blood 2012; 119:4142 - 51; http://dx.doi.org/10.1182/blood-2011-09-381970; PMID: 22408259
- Petrovic S, Leskovac A, Kotur-Stevuljevic J, Joksic J, Guc-Scekic M, Vujic D, et al. Gender-related differences in the oxidant state of cells in Fanconi anemia heterozygotes. Biol Chem 2011; 392:625 - 32; http://dx.doi.org/10.1515/bc.2011.064; PMID: 21619480
- Takeuchi T, Morimoto K. Increased formation of 8-hydroxydeoxyguanosine, an oxidative DNA damage, in lymphoblasts from Fanconi’s anemia patients due to possible catalase deficiency. Carcinogenesis 1993; 14:1115 - 20; http://dx.doi.org/10.1093/carcin/14.6.1115; PMID: 8389671
- Mukhopadhyay SS, Leung KS, Hicks MJ, Hastings PJ, Youssoufian H, Plon SE. Defective mitochondrial peroxiredoxin-3 results in sensitivity to oxidative stress in Fanconi anemia. J Cell Biol 2006; 175:225 - 35; http://dx.doi.org/10.1083/jcb.200607061; PMID: 17060495
- Kumari U, Ya Jun W, Huat Bay B, Lyakhovich A. Evidence of mitochondrial dysfunction and impaired ROS detoxifying machinery in Fanconi Anemia cells. Oncogene 2013; http://dx.doi.org/10.1038/onc.2012.583; PMID: 23318445